

- Theoretical Neuroscience Research, LLC, Ridgeland, MS, United States.
DOI:10.25259/SNI_361_2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Russell L. Blaylock. Accelerated cancer aggressiveness by viral oncomodulation: New targets and newer natural treatments for cancer control and treatment. 11-Oct-2019;10:199
How to cite this URL: Russell L. Blaylock. Accelerated cancer aggressiveness by viral oncomodulation: New targets and newer natural treatments for cancer control and treatment. 11-Oct-2019;10:199. Available from: http://surgicalneurologyint.com/surgicalint-articles/9697/
Date of Submission
17-Jun-2019
Date of Acceptance
19-Jun-2019
Date of Web Publication
11-Oct-2019
Abstract
An infectious etiology for a number of cancers has been entertained for over 100 years and modern studies have confirmed that a number of viruses are linked to cancer induction. While a large number of viruses have been demonstrated in a number of types of cancers, most such findings have been dismissed in the past as opportunistic infections, especially with persistent viruses with high rates of infectivity of the world’s populations. More recent studies have clearly shown that while not definitely causing these cancers, these viruses appear capable of affecting the biology of these tumors in such a way as to make them more aggressive and more resistant to conventional treatments. The term oncomodulatory viruses has been used to describe this phenomenon. A number of recent studies have shown a growing number of ways these oncomodulatory viruses can alter the pathology of these tumors by affecting cell-signaling, cell metabolism, apoptosis mechanisms, cell-cell communication, inflammation, antitumor immunity suppression, and angiogenesis. We are also learning that much of the behavior of tumors depends on cancer stem cells and stromal cells within the tumor microenvironment, which participate in extensive, dynamic crosstalk known to affect tumor behavior. Cancer stem cells have been found to be particularly susceptible to infection by human cytomegalovirus. In a number of studies, it has been shown that while only a select number of cells are actually infected with the virus, numerous viral proteins are released into cancer and stromal cells in the microenvironment and these viral proteins are known to affect tumor behavior and aggressiveness.
Keywords: Cytomegalovirus, Microenvironment, Oncomodulation, Viral proteins
INTRODUCTION
The cell and carcinogenic transformation
The cells in a multicellular organism possess a massive number of systems to ensure not only the survival of individual cells but also the organism as a whole [
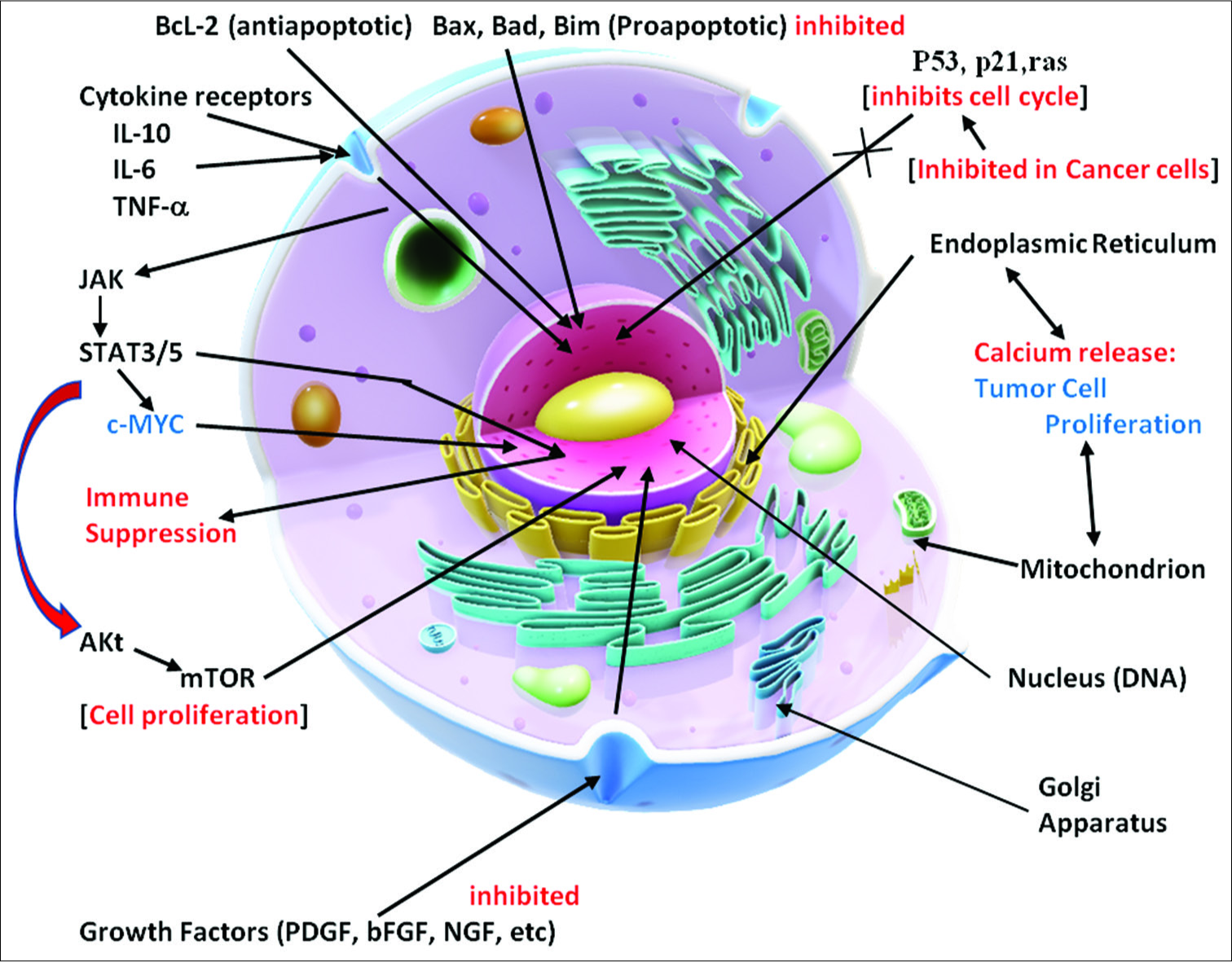
The metabolism of the cell entails a coordinated system of nutrient breakdowns, biosynthesis of essential molecules, and selection of metabolites to be utilized on an ongoing basis.
Cells also need protection from the toxic substances in its environment and need to maintain their physical structure, metabolic functions, and other maintenance requirements as the microenvironment changes. During development, and even adult maintenance, a progenitor cell may adapt to become a skin cell, a neuron, or a bone cell or an inflammatory cell from its library of contained cell instructions.
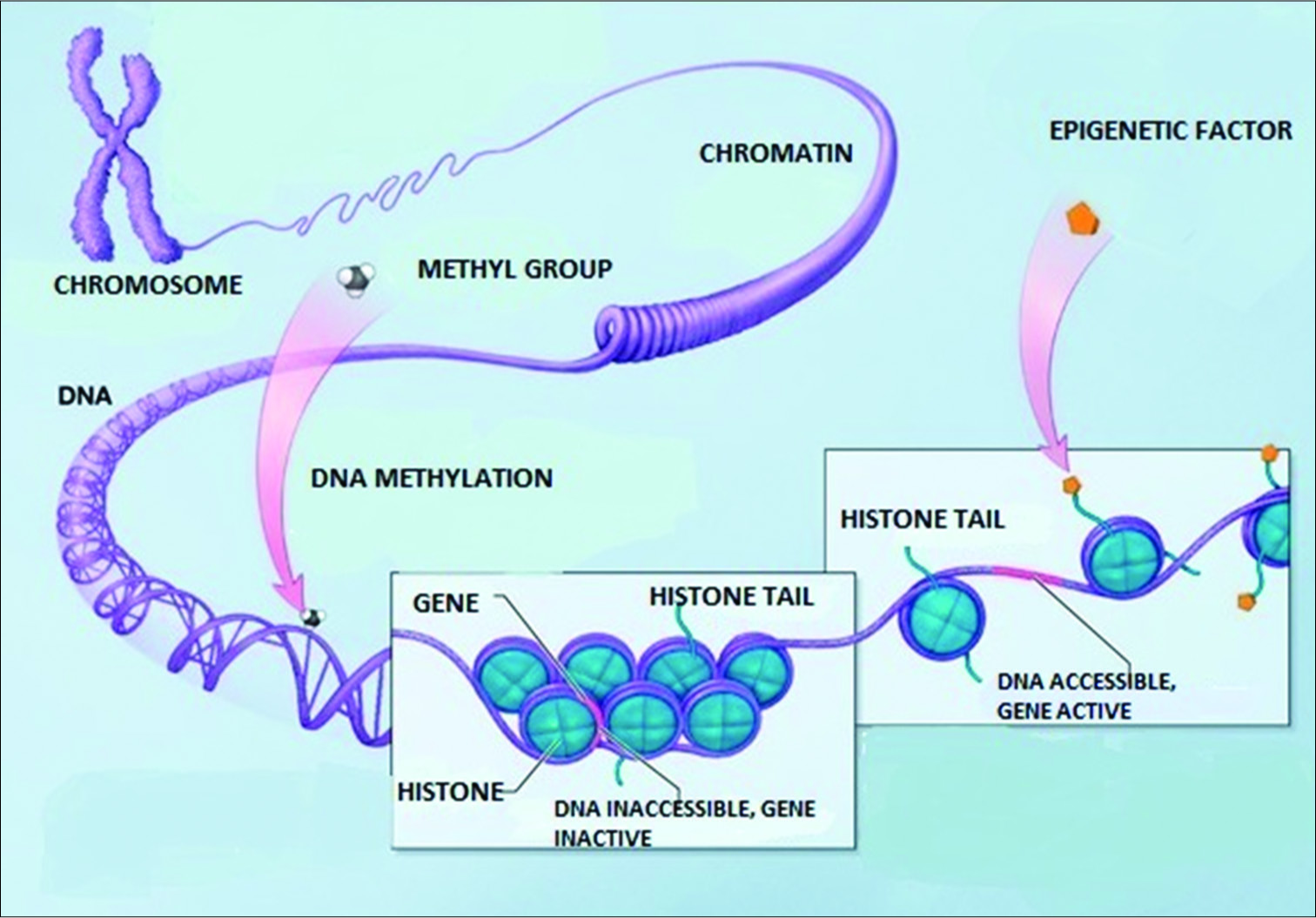
The nucleus of the cell contains its inherited library of genetic information in its DNA template. This information is stored in the DNA in its coding and noncoding sections and in the histones around which the DNA is wrapped. Specialized proteins are constructed in the nucleus on membranes in the large nuclear organelle, the nucleolus. These proteins are constructed as directed by the information contained in the nuclear library. The biochemical reactions needed for the cell to survive are selected from information in a molecular form that comes from the cellular microenvironment. All of these processes require energy sources derived from molecular reactions primarily located in mitochondrial DNA and supplied by nutrients or even in extreme circumstances from its own molecules to survive and to function. The nuclear membrane has channels to communicate with the cytosol. That is the nuclear membrane specifically allows the transport of selected molecules in and of the nucleus, especially the mRNA or message templates from the nuclear DNA that direct the cell’s various functions in the cytoplasm as well as receiving instructional transcription molecules from sites within the cytosol. The DNA itself is dynamically influenced by epigenetic signals (developmental factors in utero and during childhood, environmental chemicals, drugs, pharmaceuticals, aging, diet, and other environmental influences.) that can rapidly alter the instructions being given the cell [
The cytoplasm, or cytosol, contains water, amino acids, and organelles for the construction of proteins, which come from information derived from the nucleus. These proteins are made on short templates that come from the nucleus as mRNA protein molecules and attach to cytoplasmic membranes on the ribosomes located on the Golgi apparatus. In the cytosol, glucose, and other energy molecules are broken down or catabolized to supply energy to operate these and other metabolic processes.
The mitochondria, which contain their own genetic information, provide a major source of energy from metabolites, like pyruvate, which is formed during the process of glycolysis in the cytoplasm. Pyruvate and other metabolites enter the mitochondrion and are broken down and enter the mitochondrial tricarboxylic acid (TCA) cycle to produce high yields of energy molecules such as adenosine triphosphate (ATP). These energy molecules drive all the energy-demanding biochemical reactions, many membrane transporters, and other such functions taking place in the cell. Much of the cell’s protection is energy dependent.
Cell-signaling pathways consist of a complex series of interacting molecules within the cytoplasm and are activated by molecules from various extracellular and membrane receptors stimulated and influenced by both extracellular and intracellular signals. These extracellular activating molecules drive the cell to respond by affecting cell signaling pathways and influencing metabolism. Some signaling pathways are designed to trigger cell death (apoptosis) and others can stimulate the cells to survive, grow, and proliferate. It has been generally held that mutations in the nuclear DNA drive the cell to become a cancer cell. However, this long-held concept is now being challenged. As a result, a major change in our thinking about the origins and treatment of cancer is occurring.
Compelling evidence from multiple lines of the study suggests that most cancers are either the result of chronic inflammation or made more aggressive and deadlier as a result of prolonged and/or intense inflammation.[
Is cancer a genetic and/or a metabolic disorder?
Mechanisms linking inflammation with all stages of cancer development have been elucidated in a number of recent studies.[
Among his many studies, Seyfried has shown that cells, on malignant transformation, rapidly undergo radical changes in metabolism that favor both (a) an elevation in energy supplies and how this energy is generated and (b) rerouting of metabolism for biosynthesis of macromolecules needed for cell reproduction. To accomplish these changes requires major metabolic shifts commandeering cystolic and mitochondrial processes linked to metabolism, principally by switching metabolism to (i) favor glycolysis that takes place not only in the cytoplasm but also in the mitochondria and a (ii) redirecting the TCA cycle in the mitochondria for biosynthesis of macromolecules, production of nucleotides, and supplying substrates for membrane lipids and TCA cycle intermediates, in addition to making the general fuel source of metabolism, molecular ATP [
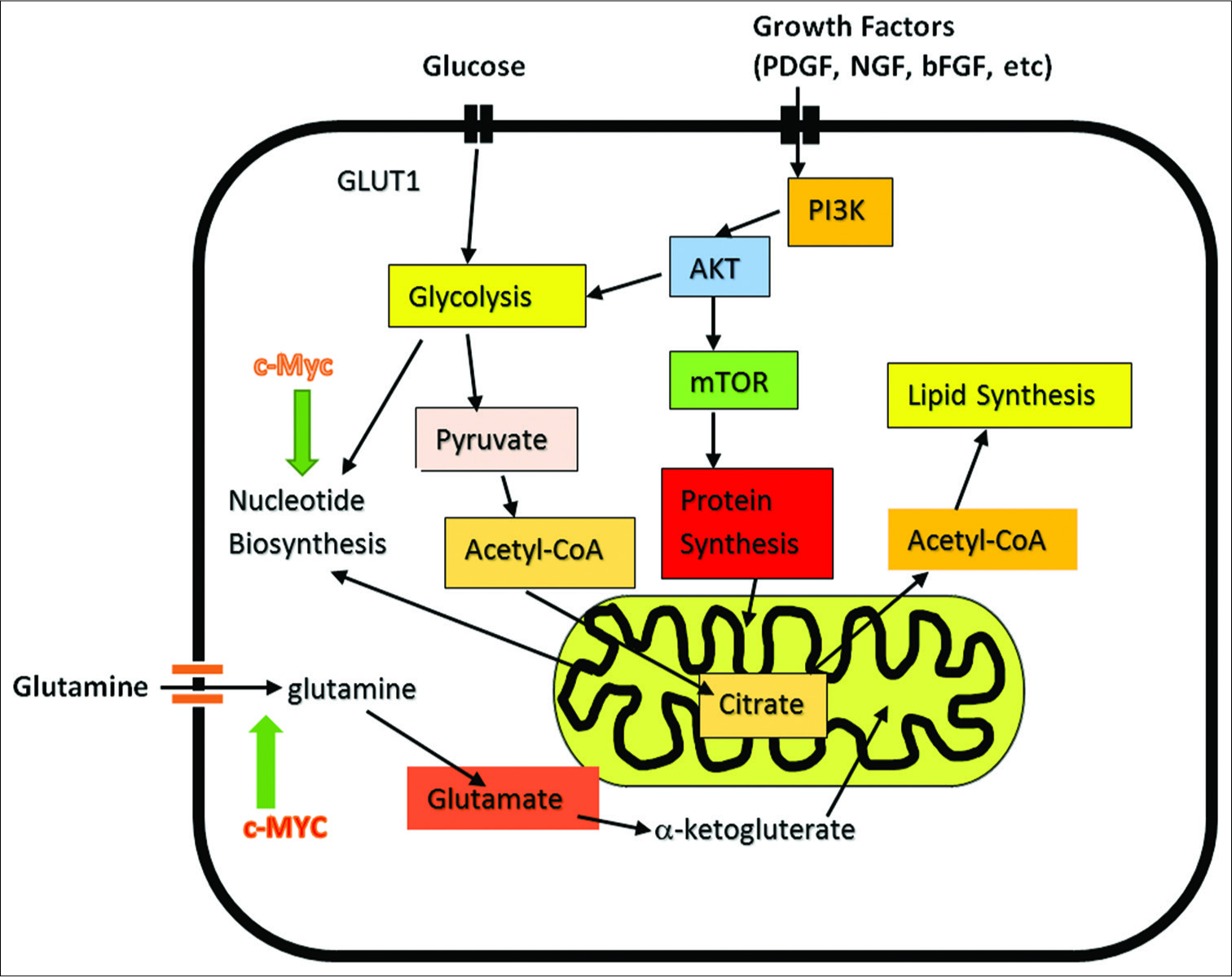
Figure 3:
Cancer Cell Metabolism involving its two major fuels, glucose and glutamine. Demonstrates role of c-MYC in induction of glutamine addiction. Myc consist of a family of regulator genes (proto-oncogenes) coding for transcription factors. The Myc family consists of c-myc, l-myc and n-myc. Also demonstrates the influence of growth factors on cancer cell-signaling.
The cancer process is also dependent on suppressing apoptosis (programmed cell death), inhibiting tumor suppressor mechanisms (p53, p21, and PTEN), and stimulating cell growth factors (Bcl-2, platelet-derived growth factors [PDGFs], basic fibroblastic growth factor, etc.) from the mitochondria [
One of the unanswered questions is - How would a cancer cell, by random mutations, know which oncogenes were essential for its survival, eventual invasion of surrounding tissues, massive proliferation of cells, activation of a complex process, and finely tuned processes, such as the angiogenesis programs, and, in an exacting way, create metabolic alterations favorable to its growth and spread? Random mutations caused from damage by reactive oxygen and nitrogen species, one would think, would activate genes that were not only favorable to tumor growth but also equally be unfavorable, being that reactive oxygen and nitrogen genetic injury would be rather haphazard.
The actual conversion of a precancer cell into a fully cancerous cell appears to involve a series of steps that must be rather exacting so as to turn on cell survival mechanisms and suppress programmed cell death mechanisms. This not only entails cell-signaling pathways but also alterations in metabolism, which appear to be quite dynamic as cancer progresses.
VIRAL CAUSATION OF CANCER: THE EVIDENCE GROWS STRONGER
There are some 1400 human pathogens that include 220 viruses. It is recognized that a variety of infectious agents can contribute to the carcinogenic process.[
Of greatest concern regarding cancer are those viruses that show persistence following acute infection. The most common persistent human pathogens include the herpes group of viruses, which include herpes simplex 1 and 2, cytomegalovirus, Epstein-Barr virus (EBV), herpesvirus-6, and herpesvirus-7. In the past, researchers referred to viruses linked to cancer causation as being oncogenic viruses. These viruses become oncogenic by a number of mechanisms, including induction of chronic inflammation, insertion of specific viral genes into the cell’s genome, inducing overexpression of carcinogenic cell signaling pathways, and altering cell metabolism.[
Until rather recently, viruses were classified as being capable of oncogenic transformation by the fact that they inserted their oncogenic genetic structure into the cells’ genome.[
What is oncomodulation?
Oncomodulation involves the production of a number of protein viral products in the cell’s cytoplasm that promote tumor cell invasion, proliferation, angiogenesis, immune suppression, altered expression of cell-signaling pathways, inhibition of apoptotic mechanisms, and suppression of tumor inhibitors, such as PTEN, pRb, p53, and p21.[
Armed with this information, it becomes obvious that all of these processes are fine-tuned to promote survival of the cancer cells against the cell’s and body’s defense mechanisms. One must ask how could cancer cells know exactly what cell mechanisms and cell signaling pathways would be needed to accomplish these goals and how to fine-tune them. The likelihood of this occurring by a totally random process of DNA damage initiated by a storm of reactive oxygen and nitrogen products, as stated, seems illogical. Yet, viruses are programmed to carry out a very similar series of processes within infected cells to ensure not only viral reproduction but also prolonged survival in a latent state.
The real debate is now between the idea that cancerous cells are merely favorable niches for these viruses or whether these viruses can actually induce cancer and then control its behavior. Compelling evidence suggests that oncomodulatory viruses can control the behavior of transformed cells and some evidence strongly suggests that certain viral proteins can induce cancerous transformation.[
Rather than discuss a number of persistent viruses linked to oncomodulation, I will focus on two of the most likely candidates, mostly human cytomegalovirus (HCMV) and less so herpes simplex virus type 1 (HSV-1). Other oncomodulatory viruses include SV-40, human adenovirus and human papillomavirus type 16 and 18.[
Viral mechanisms in cancer cell development
Making a link between viral infections and cancer transformation of normal cells demands some rather strict criteria as well as methodology and technology that was not available until relatively recently. In 2002, Cobbs et al. reported the presence of HCMV protein and nucleic acids in virtually all glioblastomas (GBM) they examined but not in normal brain tissue.[
Viruses (HCMV) can induce altered metabolism in stem cells
It was assumed in earlier studies, and some later studies, that for the virus to play a role, it must infect most of the tumor cells. Ranganathan et al. found in their frozen specimen studies of GBM that most of the tumor cells were not infected with the virus, rather only a select number of cells were actually infected, with other cells containing viral proteins known to affect tumor behavior.[
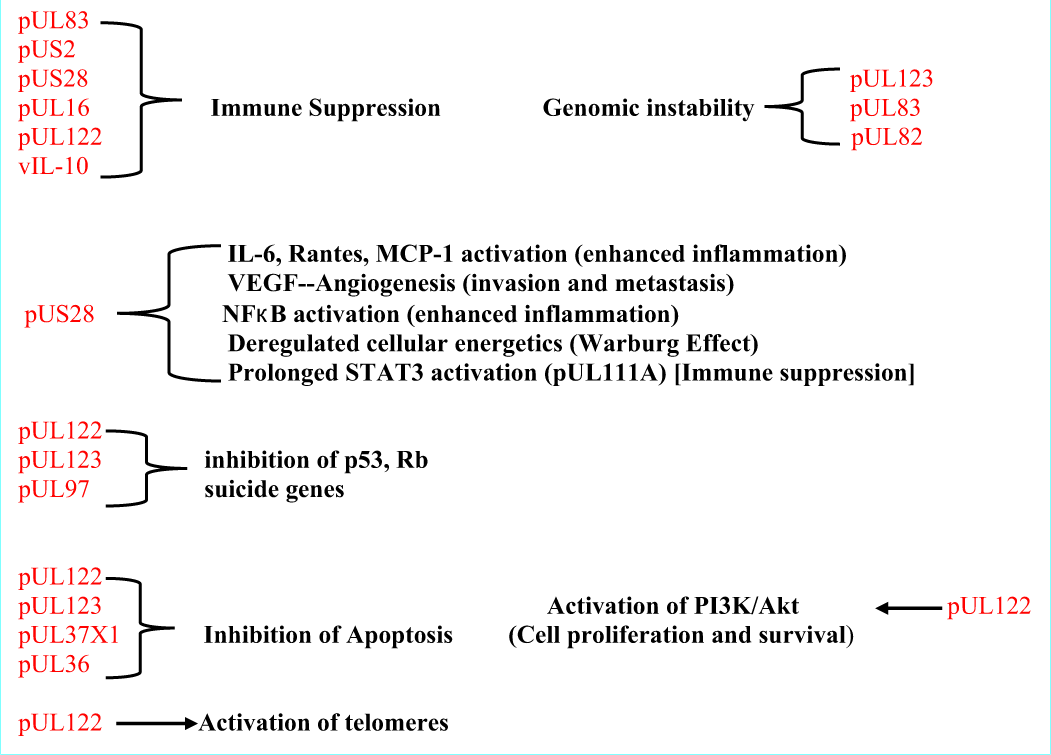
Mutated HCMV causing induction of cells to become tumors
Some have suggested that it is mutated HCMV viruses that are responsible for these findings of enhanced tumor aggressiveness. Dolan et al., for example, found that mutant forms of HCMV grew slowly in tissues, a requirement for oncomodulation.[
The latent virus may be inactive only in so far as viral reproduction and not production of the oncomodulatory proteins.[
Chronic inflammation and HCMV infection
Chronic inflammation is now considered not only a major trigger for oncogenic transformation but also drives cancer at all stages of development.[
The tumor microenvironment consists of over 50% nontumor stromal cells, which include an assortment of immune cells (tumor-associated macrophages [TAMs], dendritic cells, tumor-associated neutrophils, NK and NKT cells, B lymphocytes, and T-lymphocytes), cancer fibroblast, adipocytes, vascular endothelial cells, pericytes, and lymphatic endothelial cells.[
Cytokine interleukin (IL)-6 and activation of HCMV
Inflammation is also known to activate HCMV, and the proinflammatory cytokine IL-6 (from monocyte and macrophages) appears to play a particularly important role in its activation from a latent stage.[
High levels of IL-6 have been associated with a poor prognosis in several types of cancer [
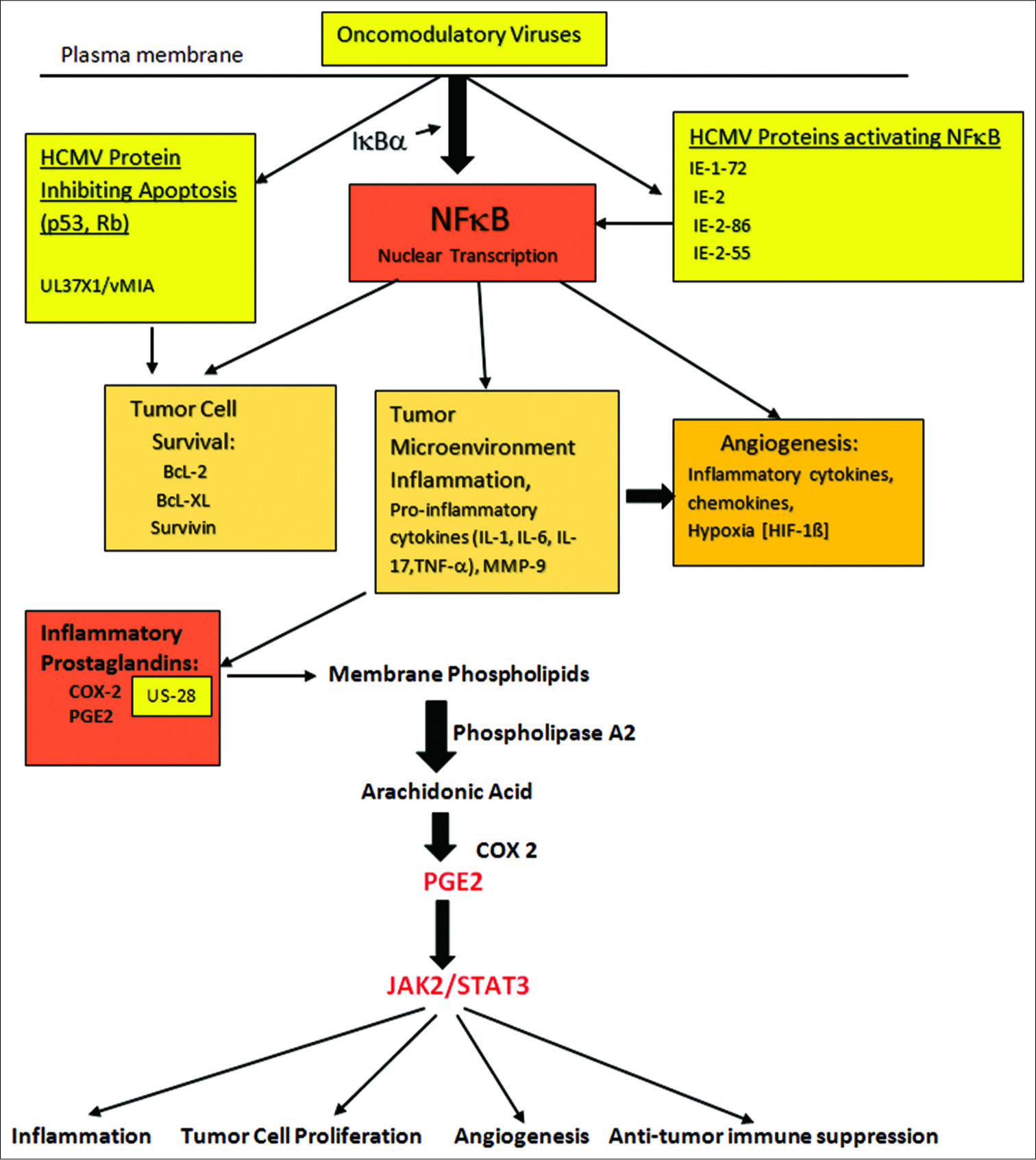
Figure 5:
Activation of COX-2 leading to generation of prostaglandins (PGE2) and subsequent enhancement of inflammation. Tumor cell proliferation, angiogenesis and suppression of anti-tumor immune activity are enhanced in the face of tumor microenvironment inflammation. The central role played by NFκB, found in all cells, is demonstrated. Activation of this factor occurs in the stromal cells, tumor cells and invading immune cells within the tumor microenvironment. PGE2 activation of JAK2/STAT3 acting on genes controlling tumor invasion/migration, cell proliferation, angiogenesis and initiating immune suppression is demonstrated.
Chemotherapy, radiotherapy, and other carcinogens in the activation of HCMV through Inflammation
It has also been shown that chemotherapy and radiotherapy can activate HCMV, which could lead to more aggressive cancers following treatment failures.[
Furthermore, of interest is the finding that the viral load of HCMV correlated with survival in glioblastoma patients. In one such study, those with low levels of the virus lived twice as long as those with the highest titers.[
In one study, nearly 80% of glioblastoma patients were found to have HCMV DNA detected in their peripheral blood, which was assumed to be from shedding of the virus by the tumor.[
Another interesting link to cancer behavior is the finding that NFκB differentially regulates the oncogene promoter c-Myc.[
The role of chronic inflammation, HCMV, and cancer cell induction
Within the brain, HCMV infection is associated with microglial activation and migration of activated macrophages to the infected brain.[
CYTOMEGALOVIRUS (HCMV) TARGETS STEM CELLS
It has been hypothesized that mutations in preneoplastic cellular tumor suppressor proteins, such as PTEN, p53, and p21, and fluctuations in the cellular microRNA profile could explain why to develop malignancies, but the most important link is to HCMV-infected stem cells.[
HCMV virus has been shown to inhibit differentiation of stem cells into neurons but not astrocytes.[
Both BMX kinase and IL-6 drive and maintain GSCs.[
Because stem cells play such a key role in the generation of cancerous tumors, as well as their maintenance and migration, the finding that HCMV preferentially infects these cells and could activate virtually all of the essential cancer cell-signaling pathways and induce critical metabolic changes within cancer stem-like cells, explains why infections of all of the cells of a tumor is not necessary for oncomodulation.
Cytomegalovirus and tumor-induced immune evasion
One of the early events in tumor development is suppression of antitumor immunity.[
Tumor microenvironment and immune cells suppressing antitumor activity
It has been observed that the tumor microenvironment generates factors that suppress antitumor immunity early in the course of the carcinogenic transition. This involves not only cancer cells but also surrounding stromal cells, which are induced by the tumor cells to release immune evading and suppressing mediators. These immune-suppressing mediators include PGE2, anti-inflammatory cytokines, chemokines, and COX-2. PGE2 interacts with nontumor cells in the tumor microenvironment, which stimulates inflammation but also suppresses antitumor immunity.[
Role of inflammation in the growth of tumors; COX-2 is a tumor growth factor and NASIDs inhibit of COX-2
It has been established that an inflammatory tumor microenvironment is crucial for sustained tumor cell proliferation, immune evasion, suppression of apoptosis mechanisms, angiogenesis, tumor invasion, and tumor cell migration. Tumor cell-induced COX-2 within the tumor microenvironment activates PGE2, which also promotes tumor growth by stimulating inflammation-driven stem cell signaling pathways essential for tumor behavior.[
Interestingly, combining antiviral drugs with COX-2 inhibitors significantly inhibited tumor growth in mice engrafted with human medulloblastoma tumors, more so than when the antiviral was used alone [
HCMV generated proteins that are immune suppressant and tumor stimulating
It has been shown that HCMV generates a number of special proteins in the tumor cell such as US28, which can bind different chemokines external to the tumor cell in its environment such as CCL2, CCL5, and CX3CL1 and thus suppressing antitumor immunity.[
HCMV US28 viral proteins have also been shown to upregulate high levels of COX-2 in HCMV infected cells.[
Studies have shown that HCMV protein expression has been detected in infiltrating inflammatory cells within the tumor microenvironment in human breast and colon cancers.[
Immune cells using IL10 cytokine and other factors to delay immune activation against the tumor; Also by HCMV
One of the more prominent immune inhibitors generated by human immune cells is IL-10, a T-helper (Th)-2 cytokine. Some pathogens use IL-10 to delay immune activation so as to establish infection.[
Through a complex interaction between tumor cells and TAMs (immunosuppressive macrophages/microglia), HCMV infections appear to impair tumor antigen presentation by dendritic cells within the tumor microenvironment.[
It is evident that HCMV controls a number of tumor cell and stromal cell mechanisms within the tumor microenvironment that can prevent antitumor immunity from functioning.
HCMV-ACTIVATED SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION (STAT) PROTEINS AND IMMUNE EVASION
STATs stimulate inflammatory response and direct antiimmune environment around the tumor
STAT family of proteins, within immune cells and tumor cells within the microenvironment, consists of seven members, STAT 1-6 and closely related STAT5A and STAT5B, which control specific biological responses [
Both NFκB and STAT3 are persistently activated in cancer cells, where they function as nuclear transcription factors required for activation of genes involved in tumor proliferation, survival, angiogenesis, immune evasion, and tumor invasion.[
STAT3 is also involved in suppressing innate antitumor immunity by promoting the conversion of antitumor macrophages into immune-suppressing TAMs and by stimulating the migration of myeloid-derived suppressor cells (MDSCs) to the tumor microenvironment.[
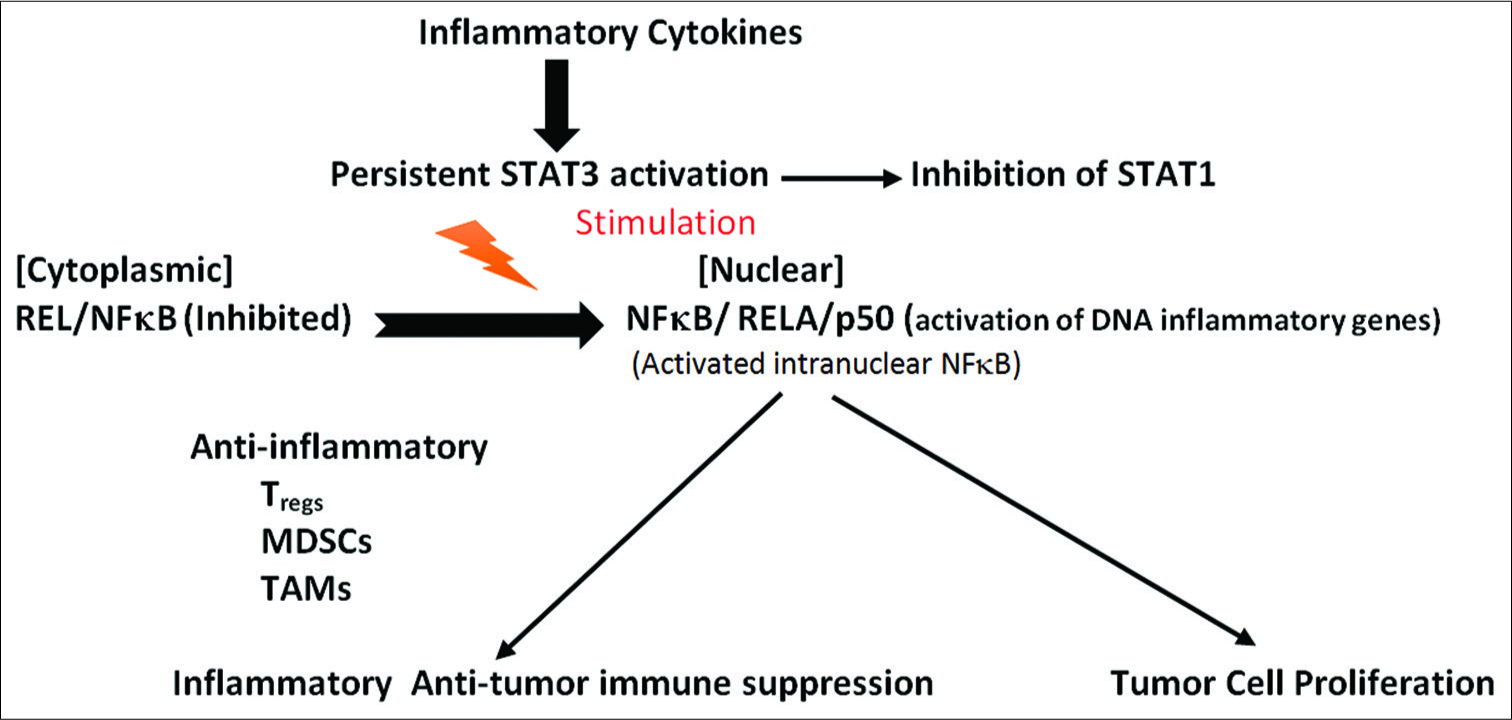
Crosstalk intercellular communication between tumor cells in the tumor microenvironment inhibiting antitumor immunity
The persistent activation of STAT3 within tumor cells crosstalks with surrounding immune cells within the tumor stroma and microenvironment to inhibit antitumor immunity.[
Activation of STAT 3 signaling by other viruses and other agents
A number of tumor viruses can activate the STAT3 pathway, including HCMV, EBV, HPV, HTLV virus, and hepatitis B virus [
How STATs influence gene regulation and apoptosis, leading to suppression of the immune response to tumors and to Stimulation of tumor cell growth and invasion mechanisms
Closely linked and critical to STAT3 pathway activation is the cytokine IL-6, downstream from NFκB.[
A number of inflammatory cytokines can activate STAT3 through NFκB/IL-6 signaling, including IL-17, IL-21, and IL- 23.[
NFκB has a dual role in cancer, acting either an anticancer pathway or procancer pathway by regulating different sets of genes.[
The procarcinogenic factor RELA is persistently activated in tumor cells and tumor-associated immune cells and requires continuous activation of STAT3.[
By activating STAT3, HCMV viruses affect a number of cancer processes including proliferation, invasion, migration, angiogenesis, and inhibition of apoptosis. Virtually, all of the alterations we see regarding immune evasion by cancers can be initiated by HCMV infections.
HCMV INHIBITION OF APOPTOSIS MECHANISMS CONTROLLED WITHIN THE MITOCHONDRIA
Cancer cells universally inhibit apoptosis by a number of mechanisms. Virtually every mechanism used to inhibit apoptosis can also be utilized by HCMV viruses, to not only suppress apoptosis but also to inhibit other tumor cell survival mechanisms. For example, in myeloma cells, IL-6 drives JAK-STAT3 pathway activation, which upregulates antiapoptotic genes [
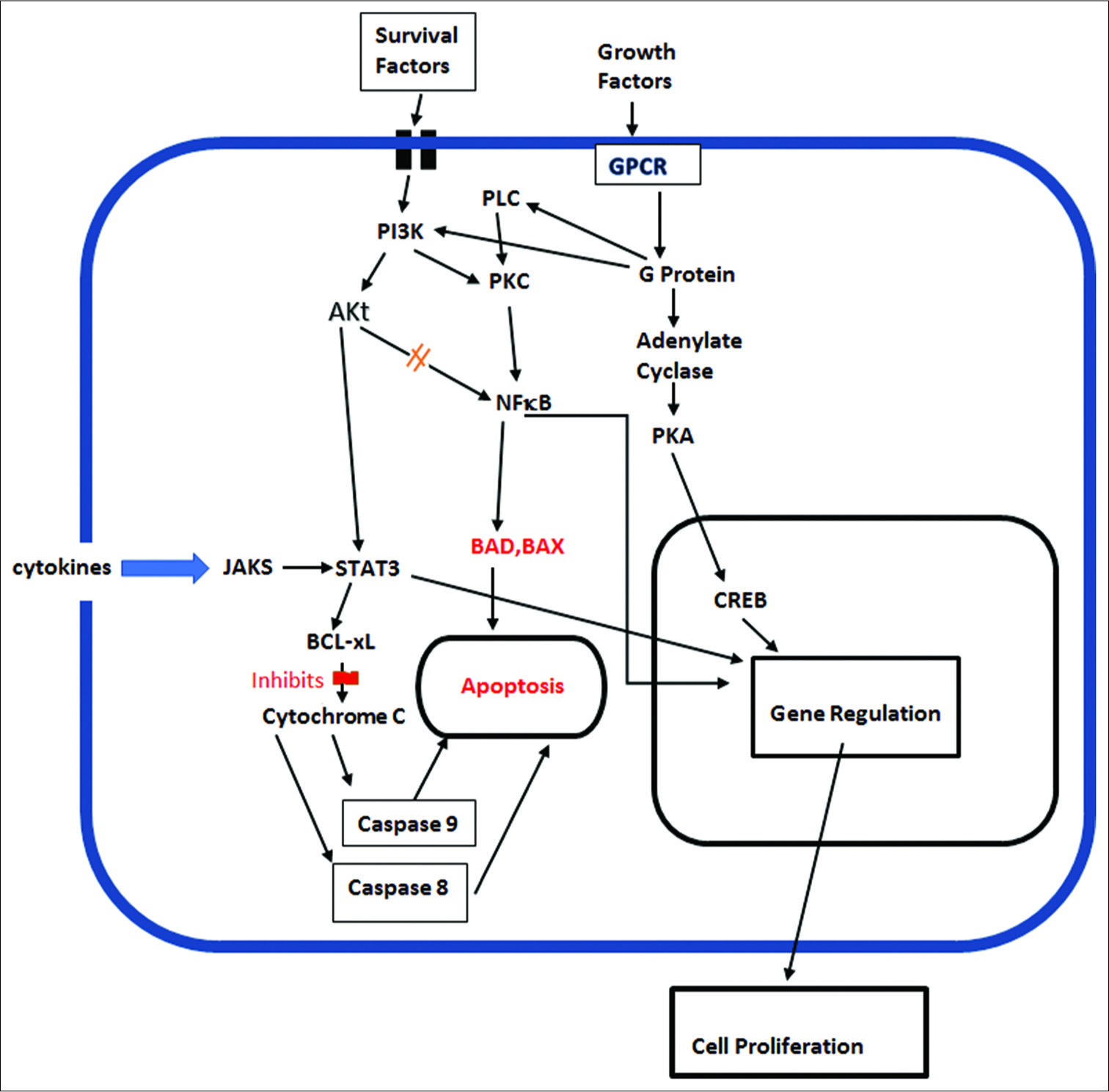
As pointed out by Williams and Colberg-Poley, viruses may encode death receptor decoys, regulate endogenous death receptor expression, direct caspase inhibitors, modulate Bcl- 2 family of proteins, or express their own viral homologous cellular Bcl-2 protein family members, to evade apoptosis.[
The major antiapoptotic members include Bcl-2 and Bcl-XL. It has been shown that HCMV-infected neuroblastoma cells were significantly less sensitive to cytotoxic anticancer drugs, such as cisplatin and etoposide, than noninfected cells.[
Other mechanisms responsible for evading cell death, such as enhancement of telomerase and a shift in error- prone DNA repair, are also playing a role in viral evasion of tumor cell apoptosis. It has been shown that HCMV induces constitutive activation of human telomerase reverse transcriptase (hTERT) in malignant glioma cells lines and most malignancies.[
Viral gene IE1 was also found to inactivate p53 and Rb tumor suppressors, while at the same time upregulating the activity of the procancer cell signaling pathway PI3-K/AKT, which enhances tumor cell survival.[
Activation of the PI3K/Akt cell signaling pathway is also important for inhibition of apoptosis in tumor cells, primarily by inhibiting the apoptosis factor BAD and restoring the antiapoptotic factors bcl-XL and bcl-2.[
TUMOR ANGIOGENESIS INDUCED BY VIRAL PROTEINS
Angiogenesis begins very early in tumor growth.[
Viral oncoproteins from several types of viruses have also been shown to stimulate the production of several angiogenic factors, including VEGF and FGF-BP, by stimulating their gene promoters in the nucleus.[
HCMV infections are characterized by a widespread presence of viruses in the vessel walls of major arteries throughout the body.[
METABOLIC ONCOMODULATION
Change from oxidative phosphorylation to aerobic glycolysis in tumor cell metabolism (Warburg effect) and the role of HCMV
Cancer cell metabolism plays a critical role in cancer cell behavior. These metabolic pathways offer a valuable target for treating and possibly curing many of the more aggressive cancers. One of the characteristics of cancer cells is a radical shift in metabolism that occurs quite early in the carcinogenic process.[
In the 1920s, Otto Warburg hypothesized that mitochondrial dysfunction was the cause of most cancers and that cancer cells switched from oxidative phosphorylation to aerobic glycolysis during malignant transformation of cells.[
One of the early events in malignant cell transformation is the dramatic increase in glucose uptake. This is accomplished in malignant cells by an increase in glucose transporters, GLUT1, and GLUT3, with higher levels of activation being associated with a poor prognosis.[
Rapidly proliferating cells have a need for accelerated macromolecule biosynthesis, such as amino acids, nucleotides, and lipids. This need is met by reducing mitochondrial respiration, which allows accumulation of intermediate precursors from glycolysis, which are then used for such synthesis, mainly through the pentose phosphate pathway [
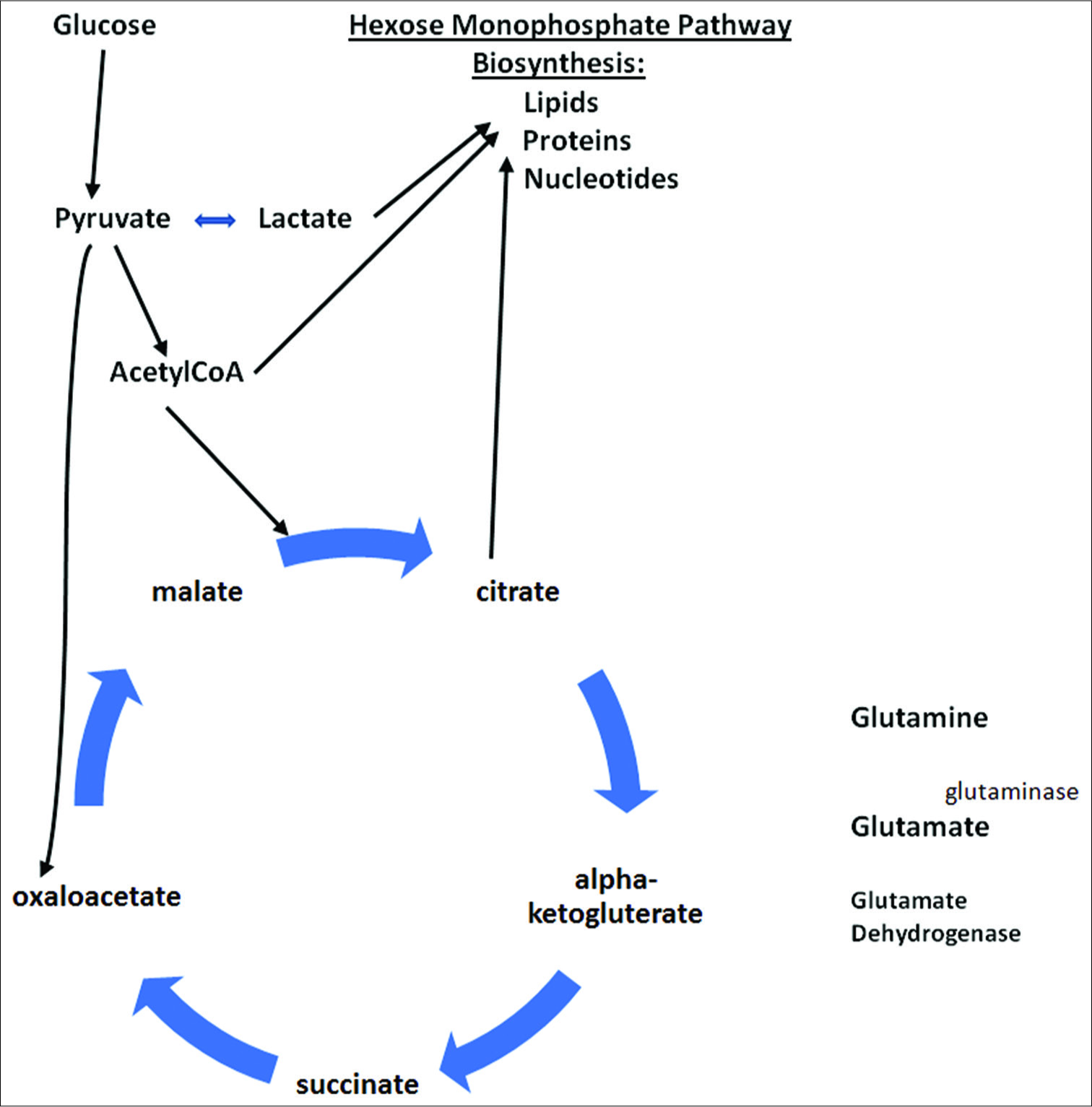
Figure 8:
Warburg effect, altering cellular metabolism to maximize use of glycolysis and Kreb’s cycle intermediates for biosynthesis of lipids, proteins and nucleotides for viral and cancer cell reproduction. Demonstrating glutaminolysis pathway for converting glutamine to glutamate and then to alpha-ketogluterate within Kreb’s cycle, used for biosynthesis of substrates.
Interestingly, HCMV has been shown to also reprogram infected cells toward a Warburg-like metabolism [
HCMV-infected cells also require an increased supply of glucose. By switching GLUT1 to GLUT4, the virus allows glucose to be transported at a rate threefold higher than can be attained by GLUT1.[
The efflux of glucose carbons from TCA cycle forms citrate, which supports fatty acid synthesis in a similar way to tumor cells.[
Glutaminolysis as a source of energy production
In
Glutamine stimulates a further accumulation of lactate (through malate formation), which increases glycolysis and NADPH generation, and this buffers oxidative stress within the cells, thus protecting the virus and the tumor cell.[
It is assumed by many researchers that the beneficial effect of glutamine metabolism for the tumor cells rests on supplying TCA cycle intermediates for biosynthesis of metabolic products, generation of glutathione and generation of NADPH required to keep glutathione in a reduced state.[
Glutamate, glutamate receptors, and tumor behavior
Circulating levels of glutamate are elevated in breast and prostate cancer patients (as well as a number of other tumor types) and high levels signify a poor prognosis.[
Further, they demonstrated that glucose depletion had no effect on invasiveness, but depletion of glutamine significantly reduced invadopodia protrusions and length. Adding glutamate to the medium restored invasive protrusions. That glutamate receptors on the invadopodia were involved in tumor invasion was demonstrated by the finding that inhibiting Group II metabotropic glutamate receptors also inhibited invasive protrusions on tumor cells. Antagonizing Groups I and II metabotropic glutamate receptors had no effect on invasiveness. Using a metabotropic glutamate receptor Group II agonist (LY40) promoted invasiveness.
Transmembrane release of matrix metalloproteinase (MMP) plays a major role in proteolytic invasion of the surrounding matrix by cancer cells. In cancer cells, MMPs becomes concentrated within the invadopodial structure.[
Tumor cells are not the only source of high levels of glutamate, as stromal fibroblast within the tumor microenvironment can also release very high levels of glutamate.[
Glutamate receptors and tumor invasiveness
Ye et al. demonstrated that high-grade pediatric gliomas released high levels of glutamate that enhanced tumor invasiveness by a process of excitotoxicity at the leading edge of the tumor.[
How the NMDA receptor antagonists are inhibiting tumor proliferation and migration is not specifically known but it may be through inhibiting ERK1/2 and CREB phosphorylation, cell signaling pathways that regulate cell proliferation.[
Failure of glutamate clearance from the tumor microenvironment
Interestingly, it has been found that glioma cell lines isolated from patient’s malignant gliomas lack the glutamate transport protein EAAT-2 (GLT-1), which is the main glutamate clearing protein in the brain.[
Calcium and tumor growth
It is known that calcium oscillations play a critical role in tumor invasion and tumor cell migration (metastasis).[
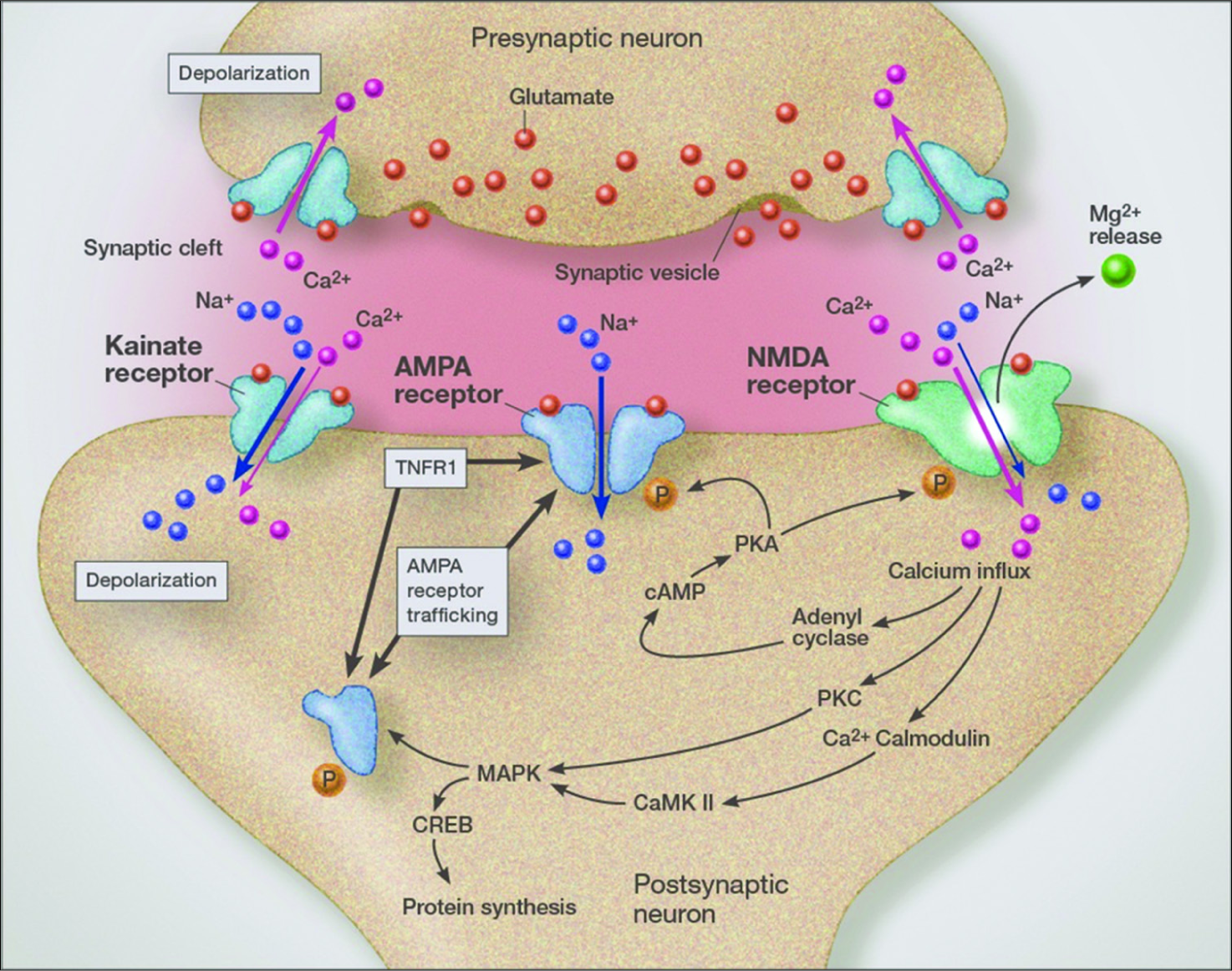


One of the important links to viral infections and other inflammatory conditions to the aggressive behavior of cancer is that inflammation increases trafficking of GluR2-lacking AMPA receptors (calcium permeable AMPA receptors) [
VACCINATIONS USED TODAY AND THEIR POTENTIAL THREAT IN THE CAUSATION OF CANCER
Of great interest is the finding that even a single viral protein, such as US28, when injected in an experimental animal can result in tumor development.[
No real effort is being made to remove these viral fragments and proteins from vaccines. With millions of people being vaccinated with these contaminated vaccines, a real and present danger of disease induction exists, including induction of cancers. Even with nononcogenic viruses, the oncomodulatory effects would have the potential to make many cancers much more aggressive and therefore deadly.
Little comfort comes from claims of no evidence of human disease from these vaccine contaminants, as most studies are not long term. If we use HCMV as a prototype, with a large percentage of people in the world carrying the virus, while it is not associated with causing cancer, as this paper shows, it has major oncomodulatory effects on established cancers.
NEUTRACEUTICALS AND THEIR ANTIVIRAL EFFECT AS A TUMOR SUPPRESSOR
It has been shown that several naturally occurring compounds, such as curcumin, quercetin, baicalin, luteolin, and resveratrol can suppress replication of a number of viruses both in vitro and in vivo.[
Grape seed proanthocyanadins extract has been shown to reduce calcium entry associated with calcium-permeable AMPA receptors, which during states of inflammation constitute a major source of intracellular calcium and calcium oscillations.[
CONCLUSION
The new finding that certain viruses, while not definitely transforming normal cells into tumor cells, can have dramatic effects on their behavior at all levels of the carcinogenic process. By an oncomodulatory series of mechanisms, viruses can make several cancers more aggressive and more likely to metastasize. The HCMV produces over 200 proteins, only a few of which have to do with viral replication - most being involved in altering host cell behavior.[
Unfortunately, despite the clear demonstration of oncomodulation by this virus and others, viral testing is not routine for cancer patients. Of even more concern, is the fact that utilizing antiviral treatments along with traditional treatments is not routine, but in my opinion, should be. A number of cited studies have shown that such combinations can significantly improve long-term survival, even for some of the more aggressive cancers, such as glioblastoma, inflammatory breast cancer, and medulloblastoma.
It appears reasonable, based on the literature, to test all cancer patients for the presence of HCMV, EBV, and HSV-1 and 2, and if found to be infected, these patients should undergo antiviral treatments - either natural or pharmaceutical.
References
1. Ahn SH, Kim HJ, Jeong I, Hong YJ, Kim MJ, Rhie DJ. Grape seed proanthocyanidin extract inhibits glutamate-induced cell death through inhibition of calcium signals and nitric oxide formation in cultured rat hippocampal neurons. BMC Neurosci. 2011. 12: 78-
2. Anzil AP, Stavrou D, Blizinger K. Type-C viral particles in cell cultures of chemically induced glioma in Sprague-Dawley rats. Vopr Onkol. 1978. 24: 30-2
3. Arena A, Liberto MC, Iannello D, Capozza AB, Focà A. Altered cytokine production after human herpes virus Type 6 infection. New Microbiol. 1999. 22: 293-300
4. Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: Cooperation of the ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006. 20: 2093-101
5. Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006. 26: 6781-90
6. Ayala FR, Rocha RM, Carvalho KC, Carvalho AL, da Cunha IW, Lourenço SV. GLUT1 and GLUT3 as potential prognostic markers for oral squamous cell carcinoma. Molecules. 2010. 15: 2374-87
7. Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow?. Lancet. 2001. 357: 539-45
8. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012. 125: 5591-6
9. Barami K. Oncomodulatory mechanisms of human cytomegalovirus in gliomas. J Clin Neurosci. 2010. 17: 819-23
10. Bardell D, Essex M. Glycolysis during early infection of feline and human cells with feline leukemia virus. Infect Immun. 1974. 9: 824-7
11. Baryawno N, Rahbar A, Wolmer-Solberg N, Taher C, Odeberg J, Darabi A. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J Clin Invest. 2011. 121: 4043-55
12. Baryawno N, Sveinbjörnsson B, Kogner P, Johnsen JI. Medulloblastoma: A disease with disorganized developmental signaling cascades. Cell Cycle. 2010. 9: 2548-54
13. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009. 8: 33-40
14. Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother. 2013. 62: 1137-48
15. Ben-Baruch A. Inflammation-associated immune suppression in cancer: The roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006. 16: 38-52
16. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011. 12: 715-23
17. Bentvelzen P. Host-virus interactions in murine mammary carcinogenesis. Biochim Biophys Acta. 1974. 355: 236-59
18. Blaylock RL, Maroon J. Natural plant products and extracts that reduce immunoexcitotoxicity-associated neurodegeneration and promote repair within the central nervous system. Surg Neurol Int. 2012. 3: 19-
19. Blaylock RL. Cancer microenvironment, inflammation and cancer stem cells: A hypothesis for a paradigm change and new targets in cancer control. Surg Neurol Int. 2015. 6: 92-
20. Boldogh I, AbuBakar S, Albrecht T. Activation of proto-oncogenes: An immediate early event in human cytomegalovirus infection. Science. 1990. 247: 561-4
21. Boldogh I, AbuBakar S, Deng CZ, Albrecht T. Transcriptional activation of cellular oncogenes fos, jun, and myc by human cytomegalovirus. J Virol. 1991. 65: 1568-71
22. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T. Gp130-mediated stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009. 15: 91-102
23. Bresnahan WA, Hultman GE, Shenk T. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J Virol. 2000. 74: 10816-8
24. Britt WJ, Mach M. Human cytomegalovirus glycoproteins. Intervirology. 1996. 39: 401-12
25. Brocke KS, Staufner C, Luksch H, Geiger KD, Stepulak A, Marzahn J. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol Ther. 2010. 9: 455-68
26. Budczies J, Pfitzner BM, Györffy B, Winzer KJ, Radke C, Dietel M. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int J Cancer. 2015. 136: 1619-28
27. Butel JS. Viral carcinogenesis: Revelation of molecular mechanisms and etiology of human disease. Carcinogenesis. 2000. 21: 405-26
28. Caposio P, Orloff SL, Streblow DN. The role of cytomegalovirus in angiogenesis. Virus Res. 2011. 157: 204-11
29. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998. 394: 485-90
30. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000. 407: 249-57
31. Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rossé C, Chavrier P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu Rev Cell Dev Biol. 2016. 32: 555-76
32. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R. Constitutive activation of stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999. 10: 105-15
33. Cavalheiro EA, Olney JW. Glutamate antagonists: Deadly liaisons with cancer. Proc Natl Acad Sci U S A. 2001. 98: 5947-8
34. Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol. 2010. 84: 1867-73
35. Chan G, Bivins-Smith ER, Smith MS, Yurochko AD. Transcriptome analysis of NF-kappaB-and phosphatidylinositol 3-kinase-regulated genes in human cytomegalovirus-infected monocytes. J Virol. 2008. 82: 1040-6
36. Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J. Disruption of stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004. 114: 720-8
37. Cheeran MC, Hu S, Ni HT, Sheng W, Palmquist JM, Peterson PK. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J Neurosci Res. 2005. 82: 839-50
38. Cheeran MC, Hu S, Yager SL, Gekker G, Peterson PK, Lokensgard JR. Cytomegalovirus induces cytokine and chemokine production differentially in microglia and astrocytes: Antiviral implications. J Neurovirol. 2001. 7: 135-47
39. Chen Z, O’Shea JJ. Th17 cells: A new fate for differentiating helper T cells. Immunol Res. 2008. 41: 87-102
40. Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J. A critical role for stat3 signaling in immune tolerance. Immunity. 2003. 19: 425-36
41. Chiou SK, Tseng CC, Rao L, White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with bcl-2 in the inhibition of apoptosis in infected cells. J Virol. 1994. 68: 6553-66
42. Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A. Analysis of the phosphatidylinositol 3’-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003. 63: 2742-6
43. Cinatl J, Bittoova M, Margraf S, Vogel JU, Cinatl J, Preiser W. Cytomegalovirus infection decreases expression of thrombospondin-1 and-2 in cultured human retinal glial cells: Effects of antiviral agents. J Infect Dis. 2000. 182: 643-51
44. Cinatl J, Cinatl J, Vogel JU, Kotchetkov R, Driever PH, Kabickova H. Persistent human cytomegalovirus infection induces drug resistance and alteration of programmed cell death in human neuroblastoma cells. Cancer Res. 1998. 58: 367-72
45. Cinatl J, Cinatl J, Vogel JU, Rabenau H, Kornhuber B, Doerr HW. Modulatory effects of human cytomegalovirus infection on malignant properties of cancer cells. Intervirology. 1996. 39: 259-69
46. Cinatl J, Nevels M, Paulus C, Michaelis M. Activation of telomerase in glioma cells by human cytomegalovirus: Another piece of the puzzle. J Natl Cancer Inst. 2009. 101: 441-3
47. Cinatl J, Vogel JU, Kotchetkov R, Wilhelm Doerr H. Oncomodulatory signals by regulatory proteins encoded by human cytomegalovirus: A novel role for viral infection in tumor progression. FEMS Microbiol Rev. 2004. 28: 59-77
48. Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res. 1995. 55: 727-30
49. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002. 62: 3347-50
50. Cobbs CS, Soroceanu L, Denham S, Zhang W, Kraus MH. Modulation of oncogenic phenotype in human glioma cells by cytomegalovirus IE1-mediated mitogenicity. Cancer Res. 2008. 68: 724-30
51. Cobbs CS. Evolving evidence implicates cytomegalovirus as a promoter of malignant glioma pathogenesis. Herpesviridae. 2011. 2: 10-
52. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002. 420: 860-7
53. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: A magic bullet?. Science. 2013. 339: 286-91
54. Dalwadi H, Krysan K, Heuze-Vourc’h N, Dohadwala M, Elashoff D, Sharma S. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin Cancer Res. 2005. 11: 7674-82
55. Darnell JE Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002. 2: 740-9
56. de Groot JF, Liu TJ, Fuller G, Yung WK. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005. 65: 1934-40
57. de la Iglesia N, Puram SV, Bonni A. STAT3 regulation of glioblastoma pathogenesis. Curr Mol Med. 2009. 9: 580-90
58. De Leo A, Arena G, Lacanna E, Oliviero G, Colavita F, Mattia E. Resveratrol inhibits epstein barr virus lytic cycle in burkitt’s lymphoma cells by affecting multiple molecular targets. Antiviral Res. 2012. 96: 196-202
59. de Wit RH, Mujić-Delić A, van Senten JR, Fraile-Ramos A, Siderius M, Smit MJ. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1α/PKM2 axis in glioblastoma cells. Oncotarget. 2016. 7: 67966-85
60. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007. 104: 19345-50
61. Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A. 2003. 100: 6801-6
62. Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res. 2010. 70: 4260-4
63. Din FV, Theodoratou E, Farrington SM, Tenesa A, Barnetson RA, Cetnarskyj R. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut. 2010. 59: 1670-9
64. Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C. Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004. 85: 1301-12
65. Dornier E, Rabas N, Mitchell L, Novo D, Dhayade S, Marco S. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat Commun. 2017. 8: 2255-
66. Dumortier J, Streblow DN, Moses AV, Jacobs JM, Kreklywich CN, Camp D. Human cytomegalovirus secretome contains factors that induce angiogenesis and wound healing. J Virol. 2008. 82: 6524-35
67. Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012. 14: 246-55
68. Dziurzynski K, Wei J, Qiao W, Hatiboglu MA, Kong LY, Wu A. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin Cancer Res. 2011. 17: 4642-9
69. Erlandsson A, Brännvall K, Gustafsdottir S, Westermark B, Forsberg-Nilsson K. Autocrine/paracrine platelet-derived growth factor regulates proliferation of neural progenitor cells. Cancer Res. 2006. 66: 8042-8
70. Evers DL, Chao CF, Wang X, Zhang Z, Huong SM, Huang ES. Human cytomegalovirus-inhibitory flavonoids: Studies on antiviral activity and mechanism of action. Antiviral Res. 2005. 68: 124-34
71. Evers DL, Wang X, Huong SM, Huang DY, Huang ES. 3,4’,5-trihydroxy-trans-stilbene (resveratrol) inhibits human cytomegalovirus replication and virus-induced cellular signaling. Antiviral Res. 2004. 63: 85-95
72. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006. 9: 425-34
73. Fenske W, Völker HU, Adam P, Hahner S, Johanssen S, Wortmann S. Glucose transporter GLUT1 expression is an stage-independent predictor of clinical outcome in adrenocortical carcinoma. Endocr Relat Cancer. 2009. 16: 919-28
74. Fiallos E, Judkins J, Matlaf L, Prichard M, Dittmer D, Cobbs C. Human cytomegalovirus gene expression in long-term infected glioma stem cells. PLoS One. 2014. 9: e116178-
75. Folkman J, Browder T, Palmblad J. Angiogenesis research: Guidelines for translation to clinical application. Thromb Haemost. 2001. 86: 23-33
76. Fortunato EA, McElroy AK, Sanchez I, Spector DH. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 2000. 8: 111-9
77. Furukawa T. A variant of human cytomegalovirus derived from a persistently infected culture. Virology. 1984. 137: 191-4
78. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004. 64: 7011-21
79. Ghandadi M, Sahebkar A. Curcumin: An effective inhibitor of interleukin-6. Curr Pharm Des. 2017. 23: 921-31
80. Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C. Latent membrane protein 1 of epstein-barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999. 18: 3064-73
81. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S. IL-6 and stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009. 15: 103-13
82. Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011. 19: 498-511
83. Hargett D, Shenk TE. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A. 2010. 107: 20039-44
84. Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet. 2002. 360: 1557-63
85. Harkins LE, Matlaf LA, Soroceanu L, Klemm K, Britt WJ, Wang W. Detection of human cytomegalovirus in normal and neoplastic breast epithelium. Herpesviridae. 2010. 1: 8-
86. Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998. 334: 297-314
87. Hendrix MG, Dormans PH, Kitslaar P, Bosman F, Bruggeman CA. The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients. Am J Pathol. 1989. 134: 1151-7
88. Ho HH, Ivashkiv LB. Role of STAT3 in Type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem. 2006. 281: 14111-8
89. Iacovelli L, Arcella A, Battaglia G, Pazzaglia S, Aronica E, Spinsanti P. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J Neurosci. 2006. 26: 8388-97
90. Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H. Blockage of ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002. 8: 971-8
91. Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via akt activation. J Neurosci. 2007. 27: 7987-8001
92. Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002. 94: 3127-34
93. Ivashkiv LB. Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur J Immunol. 2011. 41: 2477-81
94. Johnsen JI, Baryawno N, Söderberg-Nauclér C. Is human cytomegalovirus a target in cancer therapy?. Oncotarget. 2011. 2: 1329-38
95. Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: Inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol. 2001. 75: 6022-32
96. Kalariti N, Pissimissis N, Koutsilieris M. The glutamatergic system outside the CNS and in cancer biology. Expert Opin Investig Drugs. 2005. 14: 1487-96
97. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006. 6: 392-401
98. Kamdje AH, Kamga PT, Simo RT, Vecchio L, Etet PF, Muller JM. Developmental pathways associated with cancer metastasis: Notch, wnt, and hedgehog. Cancer Biol Med. 2017. 14: 109-20
99. Kastan MB, Canman CE, Leonard CJ. P53, cell cycle control and apoptosis: Implications for cancer. Cancer Metastasis Rev. 1995. 14: 3-15
100. Kazuno M, Tokunaga T, Oshika Y, Tanaka Y, Tsugane R, Kijima H. Thrombospondin-2 (TSP2) expression is inversely correlated with vascularity in glioma. Eur J Cancer. 1999. 35: 502-6
101. Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 2006. 66: 5527-36
102. Kofman A, Marcinkiewicz L, Dupart E, Lyshchev A, Martynov B, Ryndin A. The roles of viruses in brain tumor initiation and oncomodulation. J Neurooncol. 2011. 105: 451-66
103. Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D. Serum glutamate levels correlate with gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin Cancer Res. 2012. 18: 5888-901
104. Kortylewski M, Kujawski M, Herrmann A, Yang C, Wang L, Liu Y. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009. 69: 2497-505
105. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S. Inhibiting stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005. 11: 1314-21
106. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T. Regulation of the IL-23 and IL-12 balance by stat3 signaling in the tumor microenvironment. Cancer Cell. 2009. 15: 114-23
107. Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am J Pathol. 2002. 161: 919-28
108. La Rosa FA, Pierce JW, Sonenshein GE. Differential regulation of the c-Myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol Cell Biol. 1994. 14: 1039-44
109. Lau SK, Chen YY, Chen WG, Diamond DJ, Mamelak AN, Zaia JA. Lack of association of cytomegalovirus with human brain tumors. Mod Pathol. 2005. 18: 838-43
110. Le MN, Chan JL, Rosenberg SA, Nabatian AS, Merrigan KT, Cohen-Solal KA. The glutamate release inhibitor riluzole decreases migration, invasion, and proliferation of melanoma cells. J Invest Dermatol. 2010. 130: 2240-9
111. Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z. Persistently activated stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009. 15: 283-93
112. Leibovici D, Grossman HB, Dinney CP, Millikan RE, Lerner S, Wang Y. Polymorphisms in inflammation genes and bladder cancer: From initiation to recurrence, progression, and survival. J Clin Oncol. 2005. 23: 5746-56
113. Lepiller Q, Abbas W, Kumar A, Tripathy MK, Herbein G. HCMV activates the IL-6-JAK-STAT3 axis in hepG2 cells and primary human hepatocytes. PLoS One. 2013. 8: e59591-
114. Li C, Allen A, Kwagh J, Doliba NM, Qin W, Najafi H. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J Biol Chem. 2006. 281: 10214-21
115. Lo M, Wang YZ, Gout PW. The x(c)-cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J Cell Physiol. 2008. 215: 593-602
116. López-Ocejo O, Viloria-Petit A, Bequet-Romero M, Mukhopadhyay D, Rak J, Kerbel RS. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene. 2000. 19: 4611-20
117. Lv Y, Lei N, Wang D, An Z, Li G, Han F. Protective effect of curcumin against cytomegalovirus infection in balb/c mice. Environ Toxicol Pharmacol. 2014. 37: 1140-7
118. Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007. 67: 9463-71
119. Ma HI, Chiou SH, Hueng DY, Tai LK, Huang PI, Kao CL. Celecoxib and radioresistant glioblastoma-derived CD133+ cells: Improvement in radiotherapeutic effects. Laboratory investigation. J Neurosurg. 2011. 114: 651-62
120. Maeda K, Nishiguchi Y, Kang SM, Yashiro M, Onoda N, Sawada T. Expression of thrombospondin-1 inversely correlated with tumor vascularity and hematogenous metastasis in colon cancer. Oncol Rep. 2001. 8: 763-6
121. Maghazachi AA. Intracellular signaling events at the leading edge of migrating cells. Int J Biochem Cell Biol. 2000. 32: 931-43
122. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008. 454: 436-44
123. Marín YE, Chen S. Involvement of metabotropic glutamate receptor 1, a G protein coupled receptor, in melanoma development. J Mol Med (Berl). 2004. 82: 735-49
124. Matlaf LA, Harkins LE, Bezrookove V, Cobbs CS, Soroceanu L. Cytomegalovirus pp71 protein is expressed in human glioblastoma and promotes pro-angiogenic signaling by activation of stem cell factor. PLoS One. 2013. 8: e68176-
125. Matsukawa A, Kudo S, Maeda T, Numata K, Watanabe H, Takeda K. Stat3 in resident macrophages as a repressor protein of inflammatory response. J Immunol. 2005. 175: 3354-9
126. Maussang D, Verzijl D, van Walsum M, Leurs R, Holl J, Pleskoff O. Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc Natl Acad Sci U S A. 2006. 103: 13068-73
127. McCarthy M, Auger D, Whittemore SR. Human cytomegalovirus causes productive infection and neuronal injury in differentiating fetal human central nervous system neuroepithelial precursor cells. J Hum Virol. 2000. 3: 215-28
128. McGivern DR, Lemon SM. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene. 2011. 30: 1969-83
129. Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe. 2014. 15: 266-82
130. Michálek J, Horvath R. High incidence of Epstein-Barr virus, cytomegalovirus and human herpesvirus 6 infections in children with cancer. BMC Pediatr. 2002. 2: 1-
131. Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G. Constitutively activated jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995. 269: 79-81
132. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008. 10: 10-8
133. Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996. 84: 371-9
134. Mocarski ES, Kemble GW, Lyle JM, Greaves RF. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc Natl Acad Sci U S A. 1996. 93: 11321-6
135. Monick MM, Geist LJ, Stinski MF, Hunninghake GW. The immediate early genes of human cytomegalovirus upregulate expression of the cellular genes myc and fos. Am J Respir Cell Mol Biol. 1992. 7: 251-6
136. Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006. 2: e132-
137. Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008. 26: 1179-86
138. Muromoto R, Ikeda O, Okabe K, Togi S, Kamitani S, Fujimuro M. Epstein-barr virus-derived EBNA2 regulates STAT3 activation. Biochem Biophys Res Commun. 2009. 378: 439-43
139. Naithani R, Huma LC, Holland LE, Shukla D, McCormick DL, Mehta RG. Antiviral activity of phytochemicals: A comprehensive review. Mini Rev Med Chem. 2008. 8: 1106-33
140. Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep. 1985. 5: 393-400
141. Nicoletti F, Arcella A, Iacovelli L, Battaglia G, Giangaspero F, Melchiorri D. Metabotropic glutamate receptors: New targets for the control of tumor growth?. Trends Pharmacol Sci. 2007. 28: 206-13
142. Odeberg J, Wolmer N, Falci S, Westgren M, Seiger A, Söderberg-Nauclér C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol. 2006. 80: 8929-39
143. Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008. 29: 628-36
144. Ogura T, Tanaka J, Kamiya S, Sato H, Ogura H, Hatano M. Human cytomegalovirus persistent infection in a human central nervous system cell line: Production of a variant virus with different growth characteristics. J Gen Virol. 1986. 67: 2605-16
145. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J Immunol. 2009. 182: 4499-506
146. Park JS, Voitenko N, Petralia RS, Guan X, Xu JT, Steinberg JP. Persistent inflammation induces gluR2 internalization via NMDA receptor-triggered PKC activation in dorsal horn neurons. J Neurosci. 2009. 29: 3206-19
147. Perlman JM, Argyle C. Lethal cytomegalovirus infection in preterm infants: Clinical, radiological, and neuropathological findings. Ann Neurol. 1992. 31: 64-8
148. Piao Y, Lu L, de Groot J. AMPA receptors promote perivascular glioma invasion via beta1 integrin-dependent adhesion to the extracellular matrix. Neuro Oncol. 2009. 11: 260-73
149. Pollock EJ, Todaro GJ. Radiation enhancement of SV40 transformation in 3T3 and human cells. Nature. 1968. 219: 520-1
150. Polster BM, Pevsner J, Hardwick JM. Viral bcl-2 homologs and their role in virus replication and associated diseases. Biochim Biophys Acta. 2004. 1644: 211-27
151. Poltermann S, Schlehofer B, Steindorf K, Schnitzler P, Geletneky K, Schlehofer JR. Lack of association of herpesviruses with brain tumors. J Neurovirol. 2006. 12: 90-9
152. Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front Pharmacol. 2011. 2: 49-
153. Porporato PE, Filigheddu N, Pedro JM, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018. 28: 265-80
154. Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005. 16: 159-78
155. Raftery MJ, Wieland D, Gronewald S, Kraus AA, Giese T, Schönrich G. Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol. 2004. 173: 3383-91
156. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol. 2012. 86: 854-64
157. Redpath S, Ghazal P, Gascoigne NR. Hijacking and exploitation of IL-10 by intracellular pathogens. Trends Microbiol. 2001. 9: 86-92
158. Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999. 17: 2941-53
159. Reeves MB, Compton T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J Virol. 2011. 85: 12750-8
160. Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol. 2001. 75: 429-38
161. Rollins BJ. Inflammatory chemokines in cancer growth and progression. Eur J Cancer. 2006. 42: 760-7
162. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP,Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet. 2011. 377: 31-41
163. Russo V, Martienssen RA, Riggs AD.editorsEpigenic Mechanism of Gene Regulation. USA: Cold Spring Harbor Laboratory Press; 1996. p. 662-
164. Rzeski W, Ikonomidou C, Turski L. Glutamate antagonists limit tumor growth. Biochem Pharmacol. 2002. 64: 1195-200
165. Sadzuka Y, Sugiyama T, Shimoi K, Kinae N, Hirota S. Protective effect of flavonoids on doxorubicin-induced cardiotoxicity. Toxicol Lett. 1997. 92: 1-7
166. Sakao-Suzuki M, Kawasaki H, Akamatsu T, Meguro S, Miyajima H, Iwashita T. Aberrant fetal macrophage/ microglial reactions to cytomegalovirus infection. Ann Clin Transl Neurol. 2014. 1: 570-88
167. Samanta M, Harkins L, Klemm K, Britt WJ, Cobbs CS. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J Urol. 2003. 170: 998-1002
168. Samavati L, Rastogi R, Du W, Hüttemann M, Fite A, Franchi L. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol Immunol. 2009. 46: 1867-77
169. Sasaki K, Zhao X, Pardee AD, Ueda R, Fujita M, Sehra S. Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol. 2008. 181: 104-8
170. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008. 116: 79-86
171. Seleme MC, Kosmac K, Jonjic S, Britt WJ. Tumor necrosis factor alpha-induced recruitment of inflammatory mononuclear cells leads to inflammation and altered brain development in murine cytomegalovirus-infected newborn mice. J Virol. 2017. 91: e01983-16
172. Semenza GL. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol Med. 2002. 8: S62-7
173. Seyfried TN, Flores RE, Poff AM, D’Agostino DP. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis. 2014. 35: 515-27
174. Seyfried TN, Mukherjee P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr Metab (Lond). 2005. 2: 30-
175. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond). 2010. 7: 7-
176. Seyfried TN.editorsCancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer. New York: John Wiley and Sons; 2012. p.
177. Shanmugam MK, Tergaonkar V, Seiti G. Role of inflammation in cancer development. Cancer Res. 2005. 65: 5720-9
178. Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun. 2010. 391: 91-5
179. Sheng S, Qiao M, Pardee AB. Metastasis and AKT activation. J Cell Physiol. 2009. 218: 451-4
180. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009. 27: 2383-92
181. Silva EM, Mariano VS, Pastrez PR, Pinto MC, Castro AG, Syrjanen KJ. High systemic IL-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS One. 2017. 12: e0181125-
182. Sinclair J, Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol. 2006. 87: 1763-79
183. Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci Signal. 2010. 3: ra58-
184. Söderberg-Nauclér C, Johnsen JI. Cytomegalovirus in human brain tumors: Role in pathogenesis and potential treatment options. World J Exp Med. 2015. 5: 1-10
185. Söderberg-Nauclér C. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer?. J Intern Med. 2006. 259: 219-46
186. Söderberg-Nauclér C. HCMV microinfections in inflammatory diseases and cancer. J Clin Virol. 2008. 41: 218-23
187. Song G, Ouyang G, Bao S. The activation of akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005. 9: 59-71
188. Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008. 105: 287-95
189. Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008. 455: 391-5
190. Soroceanu L, Cobbs CS. Is HCMV a tumor promoter?. Virus Res. 2011. 157: 193-203
191. Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res. 2011. 71: 6643-53
192. Soroceanu L, Matlaf L, Khan S, Akhavan A, Singer E, Bezrookove V. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res. 2015. 75: 3065-76
193. Speir E, Yu ZX, Ferrans VJ, Huang ES, Epstein SE. Aspirin attenuates cytomegalovirus infectivity and gene expression mediated by cyclooxygenase-2 in coronary artery smooth muscle cells. Circ Res. 1998. 83: 210-6
194. Spencer JP, Vafeiadou K, Williams RJ, Vauzour D. Neuroinflammation: Modulation by flavonoids and mechanisms of action. Mol Aspects Med. 2012. 33: 83-97
195. Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold ME. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol. 2002. 76: 1285-92
196. Stangherlin LM, Castro FL, Medeiros RS, Guerra JM, Kimura LM, Shirata NK. Human cytomegalovirus DNA quantification and gene expression in gliomas of different grades. PLoS One. 2016. 11: e0159604-
197. Steck TL, Kaufman S, Bader JP. Glycolysis in chick embryo cell cultures transformed by rous sarcoma virus. Cancer Res. 1968. 28: 1611-9
198. Stepulak A, Sifringer M, Rzeski W, Endesfelder S, Gratopp A, Pohl EE. NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proc Natl Acad Sci U S A. 2005. 102: 15605-10
199. Stöppler H, Malerczyk C, Block K, Aigner A, Czubayko F. The human papillomavirus (HPV) 16 E6 oncoprotein leads to an increase in gene expression of the angiogenic switch molecule FGF-BP in non-immortalized human keratinocytes. Oncogene. 2001. 20: 7430-6
200. Strååt K, Liu C, Rahbar A, Zhu Q, Liu L, Wolmer-Solberg N. Activation of telomerase by human cytomegalovirus. J Natl Cancer Inst. 2009. 101: 488-97
201. Studer E, Bertoni G, Candrian U. Detection and characterization of pestivirus contaminations in human live viral vaccines. Biologicals. 2002. 30: 289-96
202. Suh Y, Afaq F, Johnson JJ, Mukhtar H. A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and wnt/EGFR/NF-kappaB-signaling pathways. Carcinogenesis. 2009. 30: 300-7
203. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006. 203: 1651-6
204. Sun S, Steinberg BM. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J Gen Virol. 2002. 83: 1651-8
205. Taher C, de Boniface J, Mohammad AA, Religa P, Hartman J, Yaiw KC. High prevalence of human cytomegalovirus proteins and nucleic acids in primary breast cancer and metastatic sentinel lymph nodes. PLoS One. 2013. 8: e56795-
206. Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001. 7: 1010-5
207. Takeda K, Akira S. STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev. 2000. 11: 199-207
208. Tan WF, Lin LP, Li MH, Zhang YX, Tong YG, Xiao D. Quercetin, a dietary-derived flavonoid, possesses antiangiogenic potential. Eur J Pharmacol. 2003. 459: 255-62
209. Tanaka J, Ogura T, Iida H, Sato H, Hatano M. Inhibitors of prostaglandin synthesis inhibit growth of human cytomegalovirus and reactivation of latent virus in a productively and latently infected human cell line. Virology. 1988. 163: 205-8
210. Tang FY, Chen CY, Shyu HW, Hong S, Chen HM, Chiou YH. Resveratrol induces cell death and inhibits human herpesvirus 8 replication in primary effusion lymphoma cells. Chem Biol Interact. 2015. 242: 372-9
211. Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S. Viral nucleic acids in live-attenuated vaccines: Detection of minority variants and an adventitious virus. J Virol. 2010. 84: 6033-40
212. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015. 35: S185-98
213. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010. 10: 181-93
214. Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. Characterization of an anti-apoptotic glycoprotein encoded by kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 2002. 21: 2602-15
215. Wang L, Liu PB, Hong JS. Inhibition of microglial activation by the herbal flavonoid baicalein attenuates inflammation-mediated degeneration of dopaminergic neurons. J Neural Transm. 2005. 112: 331-47
216. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-stat3 signaling pathway. J Exp Med. 2009. 206: 1457-64
217. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927. 8: 519-30
218. Williamson CD, Colberg-Poley AM. Intracellular sorting signals for sequential trafficking of human cytomegalovirus UL37 proteins to the endoplasmic reticulum and mitochondria. J Virol. 2010. 84: 6400-9
219. Williamson CD, DeBiasi RL, Colberg-Poley AM. Viral product trafficking to mitochondria, mechanisms and roles in pathogenesis. Infect Disord Drug Targets. 2012. 12: 18-37
220. Wilson M, Gill SK, MacPherson L, English M, Arvanitis TN, Peet AC. Noninvasive detection of glutamate predicts survival in pediatric medulloblastoma. Clin Cancer Res. 2014. 20: 4532-9
221. Winland CD, Welsh N, Sepulveda-Rodriguez A, Vicini S, Maguire-Zeiss KA. Inflammation alters AMPA-stimulated calcium responses in dorsal striatal D2 but not D1 spiny projection neurons. Eur J Neurosci. 2017. 46: 2519-33
222. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008. 105: 18782-7
223. Wu CC, Fang CY, Hsu HY, Chen YJ, Chou SP, Huang SY. Luteolin inhibits epstein-barr virus lytic reactivation by repressing the promoter activities of immediate-early genes. Antiviral Res. 2016. 132: 99-110
224. Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating wnt and notch pathways. J Exp Clin Cancer Res. 2018. 37: 256-
225. Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res. 2009. 69: 7986-93
226. Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999. 59: 4383-91
227. Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004. 4: 97-105
228. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007. 7: 41-51
229. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009. 9: 798-809
230. Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol. 2011. 85: 1573-80
231. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007. 178: 93-105
232. Yurochko AD, Kowalik TF, Huong SM, Huang ES. Human cytomegalovirus upregulates NF-kappa B activity by transactivating the NF-kappa B p105/p50 and p65 promoters. J Virol. 1995. 69: 5391-400
233. Zhang HG, Grizzle WE. Exosomes and cancer: A newly described pathway of immune suppression. Clin Cancer Res. 2011. 17: 959-64
234. Zur Hausen H. The search for infectious causes of human cancers: Where and why. Virology. 2009. 392: 1-10


Dr. Miguel A. Faria
Posted October 16, 2019, 2:38 pm
Excellent paper that corroborates with laboratory data what had been observed in clinical medicine. Another great contribution of Dr. Russell L. Blaylock to the progress of clinical medicine as well as scientific oncological research!