

- Craniofacial Unit, Department of Pediatric Neurosurgery, Hôpital Necker-Enfants Malades, Paris, France
- Department of Anesthesia, Hôpital Necker-Enfants Malades, Paris, France
- Service de Biochimie, Hôpital Lariboisière, APHP, Paris, France
Correspondence Address:
Federico Di Rocco
Craniofacial Unit, Department of Pediatric Neurosurgery, Hôpital Necker-Enfants Malades, Paris, France
DOI:10.4103/sni.sni_454_17
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Nathalie Chivoret, Eric Arnaud, Kim Giraudat, Frazer O'Brien, Leslie Pamphile, Philippe Meyer, Dominique Renier, C. Collet, Federico Di Rocco. Bilambdoid and sagittal synostosis: Report of 39 cases. 11-Oct-2018;9:206
How to cite this URL: Nathalie Chivoret, Eric Arnaud, Kim Giraudat, Frazer O'Brien, Leslie Pamphile, Philippe Meyer, Dominique Renier, C. Collet, Federico Di Rocco. Bilambdoid and sagittal synostosis: Report of 39 cases. 11-Oct-2018;9:206. Available from: http://surgicalneurologyint.com/?post_type=surgicalint_articles&p=9034
Date of Submission
01-Dec-2017
Date of Acceptance
06-Dec-2017
Date of Web Publication
11-Oct-2018
Abstract
Background:Bilambdoid and sagittal synostosis (BLSS), also called “Mercedes Benz synostosis,” is a multisutural craniosynostosis that has been described as a specific entity. However, this synostotic pattern can also be found in syndromic craniostenosis. To better define this entity we reviewed our experience with bilambdoid and sagittal synostosis.
Methods:We searched our prospective database for cases of bilambdoid and sagittal synostosis among all types of craniosynostosis. Two groups were distinguished – patients with isolated BLSS and the group of syndromic craniostenosis for whom BLSS was observed at initial presentation. We reviewed the clinical findings, associated diseases, and their management specifically for isolated BLSS patients.
Results:Thirty-nine patients were diagnosed with bilambdoid and sagittal synostosis among 4250 cases of craniosynostosis treated in our department over a period of 42 years. Among them, 8 were finally diagnosed as Crouzon syndrome. Of the 31 patients identified with isolated bilambdoid and sagittal synostosis, 25 (81%) were males and 6 (19%) were females. The average age at diagnosis was 17 months. At diagnosis, 16% of the population presented with papillary edema and 58% posterior digitate impressions. Two types of craniofacial dysmorphy were observed – a pattern with narrow occiput (71% of cases) and a pattern with dolichocephaly (29% of cases). Cerebellar tonsillar herniation was the most frequently associated malformation (61% of the isolated BLSS). Surgical management evolved during the years, and several surgical techniques were used to treat patients with BLSS, including isolated biparietal vault remodeling, posterior vault remodelling, and posterior vault expansion with internal or external distraction. In some cases, a craniocervical junction decompression was also performed. The mean follow-up was 82 months (7 years). The overall mental development was within normal limits in most children, but a mental delay was found in 25%.
Conclusion:Bilambdoid and sagittal synostosis constitute an isolated entity in almost 80% of the cases, whereas in the remaining 20% it is part of a faciocraniosynostosis syndrome. Two phenotypes may be found. Early surgical management is indicated, and several techniques can be used in this heterogeneous population. A cerebellar tonsillar prolapse is present in a majority of cases.
Keywords: Bilambdoid and sagittal synostosis, Chiari malformation, complex craniosynostosis, epidemiology, surgical strategies, outcome, papilledema
INTRODUCTION
Craniosynostosis occurs in one out 2000 live births.[
In the present report, we retrospectively reviewed the complex synostosis not involving the coronal sutures, and in particular, the trisutural pattern of complex craniostenosis which presents with sagittal and bilambdoid involvement (BLSS).
Neuhauser et al. reported in 1976[
Little is known about BLSS. It seems to be a heterogeneous disease. To better define this entity, we retrospectively searched in our prospective database cases of bilambdoid and sagittal synostosis among all types of craniosynostosis. This study represents, to the best of our knowledge, the largest published series of patients with BLSS. We reviewed the clinical findings, epidemiological data, associated diseases, and management for these patients and discuss the surgical strategy.
PATIENTS AND METHODS
Population
The study was conducted among consecutive patients with confirmed BLSS diagnosed between 1972 and 2014 at Necker Enfants Malades. We reviewed the clinical records, radiological findings, and their management.
The clinical diagnosis of bilambdoid and sagittal synostosis was confirmed on X-ray, skull computed tomography (CT), and for the most recent cases three-dimensional CT images, showing bilambdoid and sagittal synostosis with open coronal sutures.
RESULTS
Among 4250 cases of craniosynostosis treated in our department, 39 patients were diagnosed with bilambdoid and sagittal synostosis (0.9% of the global population) over a period of 42 years. The mean age at presentation was 23 months (3 days to 8 years). Eighty percent of this population was male. The trisutural pattern of craniostenosis was isolated in 31 cases (almost 80% of the cases), whereas a syndromic craniostenosis was identified in 8 cases (8 patients demonstrating during follow-up a clinical Crouzon syndrome which was genetically confirmed in recent patients). Papillary oedema was present in 6 cases at diagnosis (15% of the cases).
Isolated BLSS (n = 31)
Of these 31 patients identified with isolated bilambdoid and sagittal synostosis, 25 (81%) were males and 6 (19%) were females. The mean age at diagnosis was 17 months (4 days to 88 months). The age at diagnosis was less than or equal to 6 months in 52% of the cases. However, the mean age at diagnosis decreased over the years due to better recognition of craniofacial anomalies by pediatricians and improvement in radiological investigations. This trend was also shown by the increase in the number of patients diagnosed with BLSS during time [
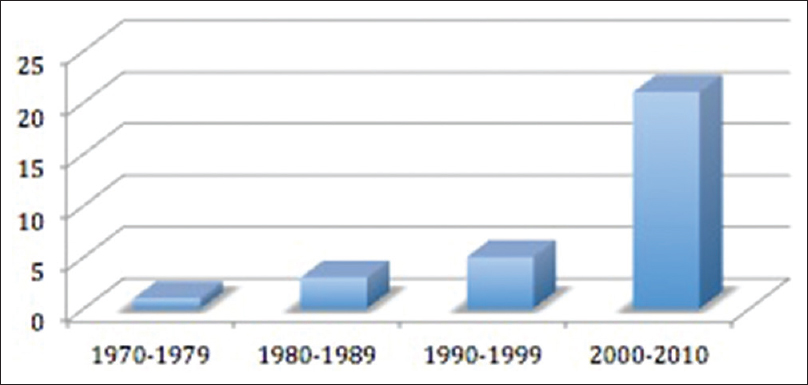
Among them, 23 patients originated from Europe, 4 from Maghreb, and 1 from Sub-Saharan Africa. For 3 patients, data were unknown. One case of consanguinity was found but no additional family members with craniosynostosis were reported.
Two patterns of cranial features could be distinguished:
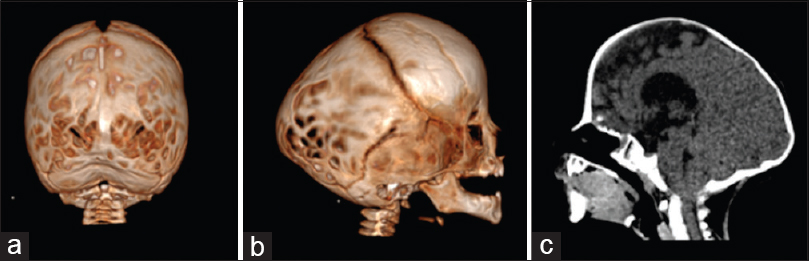
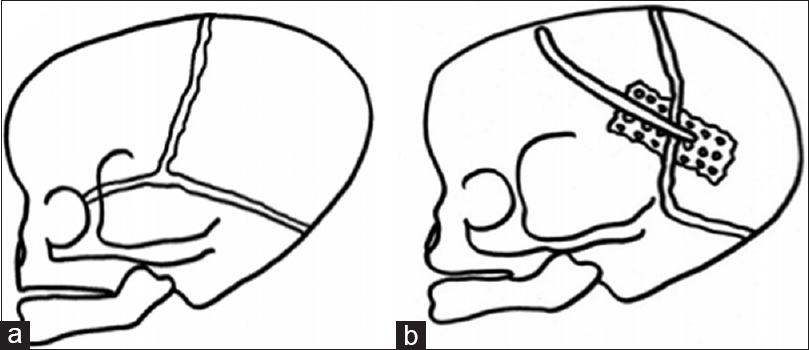
Figure 2
Three-dimensional CT showing bilateral lambdoid and sagittal synostosis with narrow occiput (“short BLSS”) in a posterior view (a) and a lateral view (b). We can notice concave occipital bone, severe posterior digitate impressions of the skull (a and b), and the descent of the cerebellar tonsils on sagittal CT scan reconstructions (c)
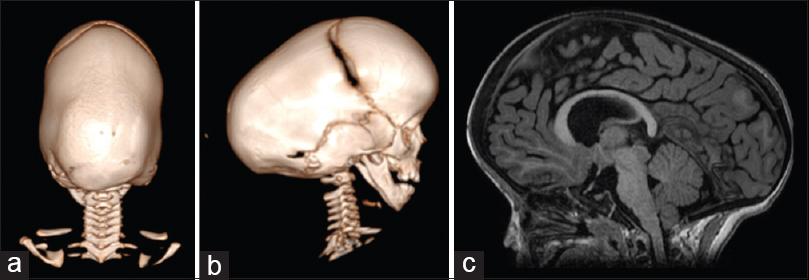
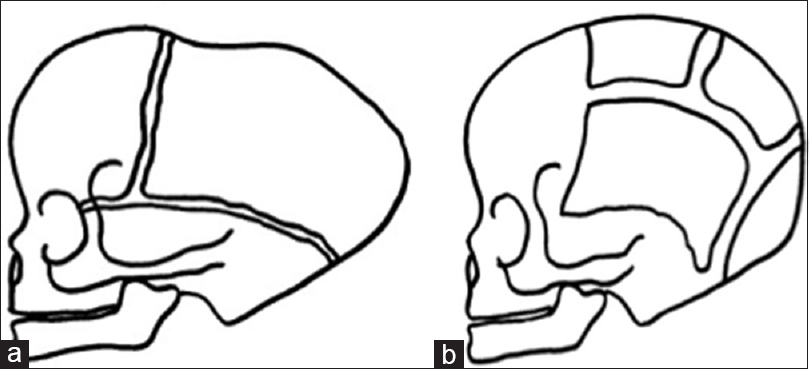
Figure 3
Three-dimensional CT scan showing a pattern of bilateral lambdoid and sagittal synostosis with dolichocephaly and occipital bossing (“long BLSS”) in a posterior view (a) and a lateral view (b) and T1 sagittal MRI exam showing a “long BLSS” pattern with moderate ventricles dilatation and small posterior fossa (c)
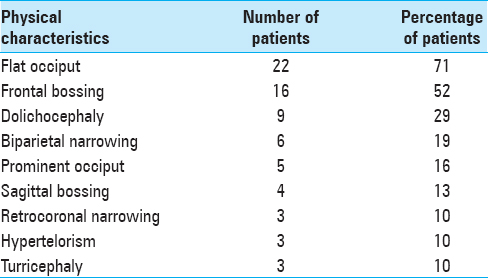
The clinical findings observed in BLSS are described in
Associated anomalies
At diagnosis, 16% of this population of BLSS presented papillary edema and 58% posterior digitate impressions. Multiple central nervous system (CNS) malformations were found in patients with BLSS. The most commonly reported were cerebellar tonsillar herniation, (19 patients; 61% of isolated BLSS population), enlarged bifrontal subarachnoid spaces (12 patients; 39% of the cases), venous anomalies (11 patients; 35% of the cases), enlargement of the cerebral ventricles (10 patients; 32% of the cases), syringomyelia (1 patient; 3% of the cases), microcephaly with bilateral deafness (1 patient; 3% of the cases), and gyration anomalies (1 patient; 3% of the cases) [
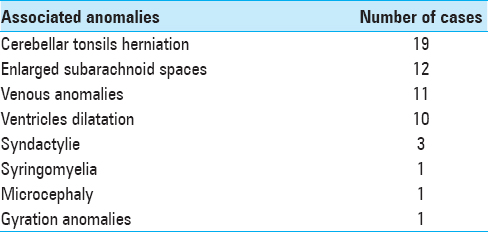
Though tonsillar herniation concerned almost two-third of the population, only 2 symptomatic cases were reported.
Similarly, though 32% of the patients presented with an enlargement of the cerebral ventricles, no cases of active hydrocephalus were found and no treatment was required.
Interestingly, the population of BLSS with cerebellar tonsillar herniation presented more commonly other anomalies such as ventricular dilatation compared to those without cerebellar tonsillar herniation. Over half of these patients with the association of ventricular dilation and cerebellar tonsillar herniation presented with symptoms that prompted a surgical decompression of the foramen magnum.
Extra-CNS malformations were also present in this population of isolated BLSS. One patient presented syndactyly of digit IV and V and low-set ears and another a brachydactyly. One patient presented multiple malformations with costal, vertebral, and urogenital malformations. Another patient presented isolated congenital urogenital malformation. No genetic syndrome was identified in these cases.
Surgical results
Of the 31 patients, 25 had reconstructive surgery (81%). The mean age at surgery was 21 months (3 months to 8 years). For 6 patients, no surgery was performed. Five patients were not operated because parents refused the treatment or because the child was considered too old due to a delayed diagnosis. In one child, due to severe hemorrhagic complication at the beginning of the procedure, the originally planned cranial remodelling was not performed and no second surgery was performed.
Two main types of cranial vault reconstruction were used in our experience – (1) posterior decompression and (2) parietal remodelling.
Posterior decompression was applied in 17 patients. All patients presented a “short BLSS,” i.e., the phenotype with occipital narrowing. In 11 patients, a parieto-occipital decompression without internal or external distraction was performed and a sagittal craniectomy was combined in 4 cases. Foramen magnum decompression was associated if tonsillar prolapse was observed (in 5 cases). For the most recent cases with occipital narrowing (6 cases), a parieto-occipital decompression with external distraction was performed without foramen magnum decompression [
Another type of cranial vault remodelling, parietal remodelling with sagittal craniectomy, was used in 8 patients [
In association with these two techniques of remodelling, a surgical decompression of the craniocervical junction was performed in 8 patients. In 6 cases, this decompression was performed during the same anesthesia for the cranial vault remodelling. In 2 cases, it was performed during a separate procedure (2 cases of symptomatic tonsillar herniation). One of these two symptomatic patients required a second procedure of craniocervical junction decompression because of development at follow-up of motor deficit and increase in the size of a syrinx cavity. This was a case of pattern of “short BLSS” with narrow occiput.
Surgical complications were observed in few patients. Dura mater tears were observed in 5 patients and local infection in one case. Major complications as venous sinus injury occurred in one case. Overall, of the 25 patients operated on, 3 required more than one surgery – 1 patient because of infection, 1 required 2 surgeries because of bad functional result after foramen magnum decompression, and finally a third patient required 3 operations due to hemorrhagic complications during the first procedure with limitation of cranial vault remodelling and because of bad aesthetic result after the second procedure.
Follow-up
The mean follow-up was 82 months (7 years). No additional synostosis was observed during the follow-up period. At the last follow-up, 8 patients (25% of patients) presented a moderate-to-severe developmental delay. In this population, 4 patients presented with a delay in diagnosis (age at diagnosis between 20 and 51 months) and one a surgical complication with notable limitation of cranial vault remodelling. Of the 4 patients with delayed diagnosis, 3 demonstrated severe signs of intracranial hypertension at diagnosis and the other microcephaly with no craniofacial surgery being performed.
Aesthetic result was considered good or excellent in 75% of the cases and insufficient in the rest of the cases. Insufficient aesthetic result required a second procedure in only one case.
Crouzon syndrome (n = 8)
Among the overall population of BLSS, a Crouzon syndrome was identified and genetically confirmed in 8 patients. Six patients were males and 2 were females. The mean age at diagnosis was 44 months (4–102). The mean paternal age was 37 years and the mean maternal age was 33 years.
DISCUSSION
BLSS is a rare pattern of complex craniostenosis. Only 39 cases were found among the population of 4250 craniosynostosis managed at Necker hospital between 1972 and 2014 (0.9%). Only 44 cases were reported in the literature.[
Interestingly, in our series, though 79% of the cases (31 patients) had an isolated BLSS, 8 children actually had a Crouzon syndrome (21%). In cases of Crouzon syndrome, no involvement of the coronal sutures was found at diagnosis or during follow-up. Because of the high incidence of this type of syndrome, a genetic testing should be considered in case of an infant with BLSS to rule out a FGFR2 mutation, allowing proper management and parental counseling.
Phenotypes
Two types of deformation can be distinguished in BLSS. The most frequent phenotype in our series was characterized by a relative brachycephaly with occipital narrowing – the “short BLSS.” A similar phenotype was observed by Neuhauser et al.[
Associated anomalies
MRI remains the optimal imaging modality in the assessment of CNS-associated anomalies in this population. The patients in this study had evidence of cerebellar tonsil's herniation, venous anomalies, and ventricular enlargement, which are perhaps related to the significant bony abnormality associated to the fusion of the lambdoid sutures.
The most frequent associated CNS anomaly reported in our series was the tonsillar herniation presenting in 61% of isolated BLSS. Such an association between tonsillar herniation and craniofacial synostosis, first described in 1972 by Saldino et al.[
One-third of the patients presented a ventricular dilatation in our series. However, no active hydrocephalus was observed and no patient required a treatment for cerebrospinal fluid (CSF) disorders. Enlarged ventricles are frequent in complex craniostenosis. Two main pathogenic factors may explain the ventricular enlargement – a mechanically increased CSF outflow resistance due to compression of posterior fossa and a raised pressure secondary to venous outflow obstruction. These pathogenic mechanisms may coexist in BLSS.[
Venous anomalies such as jugular stenosis or thrombosis, lateral sinus compression, sagittal sinus compression, or hypoplasia of lateral sinus were found in 35% of isolated BLSS in our study.
Like other multisutural craniostenosis, BLSS can be associated with intracranial hypertension. In our series, 16% of the patients presented at the time of diagnosis a papillary edema and 58% posterior digitate impressions. This is another argument to better identify this pattern of craniostenosis for an early surgical management to prevent ophthalmologic and neurologic deterioration.
Surgical management
Several surgical techniques can be used in BLSS. The surgical management may differ according to the two phenotypic patterns. Indeed in “long BLSS,” i.e. dolichocephaly-like, the goal was shortening the anteroposterior length by opening the prematurely fused suture associated with biparietal remodelling. Conversely, in “short BLSS,” i.e. brachycephaly with occipital narrowing, the goal was skull elongation by posterior decompression with or without distraction. For all recent cases, a technique of external distraction osteogenesis was used. The use of external distraction (two or three) for calvaria vault expansion is a safe and efficient method offering the important advantage of controlling the vectors of distraction, and allowing gradual expansion. Furthermore, the distractors allow the child to rest supine in the postoperative period. This technique seems to be effective to treat the group of “short BLSS” with occipital narrowing.
The question whether to open the foramen magnum or not in asymptomatic patients is still debated. Despite a preventive early opening, there is a risk of secondary stenosis and symptom appearance during follow-up. Therefore, some authors have recommend to perform surgery after 12 months of life to reduce the risk of needing a new surgical procedure for a secondary stenosis of the foramen magnum with the argument that the likelihood of dural regeneration of bone would be lower at this later age.[
Our study carries some limitations; because of the rarity of such a complex subtype, the period of the study was extremely long. Surgical and anesthetic procedures evolved during the last decades and as such reason; different techniques were applied in this population. Moreover, the rarity and heterogeneity of this complex craniostenosis hinder the possibility of randomized trials on large series to compare the different techniques.
CONCLUSION
We presented 39 cases of bilambdoid and sagittal synostosis (BLSS). This large series allow us to describe this entity and characterize the clinical features, evolution, and surgical management of this rare multisutural craniostenosis.
BLSS could be found in syndromic forms, i.e. Crouzon syndrome, underlying the importance of genetic testing. Nevertheless, in most cases in this series (31 cases), BLSS remained an isolated entity with no additional synostosis observed at follow-up analysis. Two main phenotypes can be found associated with trisutural synostosis – the “short BLSS” and the “long BLSS.” Because these children, especially when presenting with the latter phenotype, may be misdiagnosed, care must be taken to analyze the patency of the lambdoid sutures during clinical examination and on eventual radiological examinations (ultrasounds, CT, or MRI).
MRI evaluation is recommended in the management of these patients to identify associated anomalies and plan the surgical procedure. The most frequent associated anomaly is cerebellar tonsillar herniation. Venous anomalies and ventricles enlargement were also observed. Such a high incidence of tonsillar herniation in BLSS compared to other craniostenoses can be explained by the early involvement of lambdoid sutures and small posterior fossa. Early surgical correction seems to be required in this population of BLSS to try to reduce the risk of developmental delay.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Al-Torki NA, Sabry MA, Al-Tawari A, Al-Kandari NH, Al-Awadi SA. Craniofacial dyssynostosis with cryptorchidism and normal stature. Am J Med Genet. 1998. 79: 5-7
2. Bermejo E, Félix V, Lapunzina P, Galán E, Soler V, Delicado A. Craniofacial dyssynostosis: Description of the first four Spanish cases and review. Am J Med Genet A. 2005. 132A: 41-8
3. Bernardini L, Castori M, Capalbo A, Mokini V, Mingarelli R, Simi P. Syndromic craniosynostosis due to complex chromosome 5 rearrangement and MSX2 gene triplication. Am J Med Genet A. 2007. 143A: 2937-43
4. Cinalli G, Spennato P, Sainte-Rose C, Arnaud E, Aliberti F, Brunelle F. Chiari malformation in craniosynostosis. Childs Nerv Syst. 2005. 21: 889-901
5. Coll G, Arnaud E, Collet C, Brunelle F, Sainte-Rose C, Di Rocco F. Skull base morphology in fibroblast growth factor receptor type 2-related faciocraniosynostosis: A descriptive analysis. Neurosurgery. 2015. 76: 571-83
6. Coll G, Arnaud E, Selek L, Brunelle F, Sainte-Rose C, Collet C. The growth of the foramen magnum in Crouzon syndrome. Childs Nerv Syst. 2012. 28: 1525-35
7. Collmann H, Sörensen N, Krauss J. Hydrocephalus in craniosynostosis: A review. Childs Nerv Syst. 2005. 21: 902-12
8. Czerwinski M, Kolar JC, Fearon JA. Complex craniosynostosis. Plast Reconstr Surg. 2011. 128: 955-61
9. Di Rocco F, Arnaud E, Meyer P, Sainte-Rose C, Renier D. Focus session on the changing “epidemiology” of craniosynostosis (comparing two quinquennia: 1985-1989 and 2003-2007) and its impact on the daily clinical practice: A review from Necker Enfants Malades. Childs Nerv Syst. 2009. 25: 807-11
10. Fitzpatrick DR. Filling in the gaps in cranial suture biology. Nat Genet. 2013. 45: 231-2
11. Grosso S, Vivarelli R, Muraca MC, Berardi R, Marconcini S, Morgese G. Craniofacial dyssynostosis: Case report and review. Am J Med Genet A. 2004. 129A: 300-2
12. Hayward R. Venous hypertension and craniosynostosis. Childs Nerv Syst. 2005. 21: 880-8
13. Hing AV, Click ES, Holder U, Seto ML, Vessey K, Gruss J. Bilateral lambdoid and sagittal synostosis (BLSS): A unique craniosynostosis syndrome or predictable craniofacial phenotype?. Am J Med Genet A. 2009. 149A: 1024-32
14. Lahidji SF, Buchman SR, Muraszko K, Innis JW, Keegan CE. Craniofacial dyssynostosis in two boys with apparently normal cognitive development. Am J Med Genet A. 2006. 140: 1333-6
15. Moore MH, Abbott AH, Netherway DJ, Menard R, Hanieh A. Bilambdoid and posterior sagittal synostosis: The Mercedes Benz syndrome. J Craniofac Surg. 1998. 9: 417-22
16. Morton JE. Craniofacial dyssynostosis: A further case report. Am J Med Genet. 1998. 79: 8-11
17. Neuhäuser G, Kaveggia EG, Opitz JM. Studies of malformation syndromes of man XXXIX: A craniosynostosis-craniofacial dysostosis syndrome with mental retardation and other malformations: “Craniofacial dyssynostosis”. Eur J Pediatr. 1976. 123: 15-28
18. Renier D, Lajeunie E, Arnaud E, Marchac D. Management of craniosynostoses. Childs Nerv Syst. 2000. 16: 645-58
19. Rhodes JL, Kolar JC, Fearon JA. Mercedes Benz pattern craniosynostosis. Plast Reconstr Surg. 2010. 125: 299-304
20. Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JAC, Hoogeboom AJM. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1X, are a frequent cause of coronal craniosynostosis. Nat Genet. 2013. 45: 304-7
21. Saldino RM, Steinbach HL, Epstein CJ. Familial acrocephalosyndactyly (Pfeiffer syndrome). Am J Roentgenol Radium Ther Nucl Med. 1972. 116: 609-22
22. Tamburrini G, Caldarelli M, Massimi L, Gasparini G, Pelo S, Di Rocco C. Complex craniosynostoses: A review of the prominent clinical features and the related management strategies. Childs Nerv Syst. 2012. 28: 1511-23
23. Vinchon M, Pellerin P, Baroncini M, Wolber A, Dhellemmes P. Non-syndromic oxycephaly and brachycephaly: A review. Childs Nerv Syst. 2012. 28: 1439-46

