

- Department of Neurosurgery, Hospital São Francisco, Ribeirão Preto, São Paulo, Brazil
- Department of Radiology, Hospital São Francisco, Ribeirão Preto, São Paulo, Brazil
- Department of Internal Medicine, Hospital São Francisco, Ribeirão Preto, São Paulo, Brazil
Correspondence Address:
Breno Nery
Department of Internal Medicine, Hospital São Francisco, Ribeirão Preto, São Paulo, Brazil
DOI:10.4103/sni.sni_41_18
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Breno Nery, Leandro César Tângari Pereira, Rodrigo Antônio Fernandes Costa, Rodolfo Mendes Queiroz, Lucas Giansante Abud, Eduardo Quaggio, Lígia Henriques Coronatto, Isadora Salviano Teixeira Prado, Cecília Hissae Miyake, Fred Bernardes Filho. Cervicomedullary primitive neuroectodermal tumor of the spine: Case report. 28-Nov-2018;9:241
How to cite this URL: Breno Nery, Leandro César Tângari Pereira, Rodrigo Antônio Fernandes Costa, Rodolfo Mendes Queiroz, Lucas Giansante Abud, Eduardo Quaggio, Lígia Henriques Coronatto, Isadora Salviano Teixeira Prado, Cecília Hissae Miyake, Fred Bernardes Filho. Cervicomedullary primitive neuroectodermal tumor of the spine: Case report. 28-Nov-2018;9:241. Available from: http://surgicalneurologyint.com/surgicalint-articles/9093/
Date of Submission
31-Jan-2018
Date of Acceptance
23-Oct-2018
Date of Web Publication
28-Nov-2018
Abstract
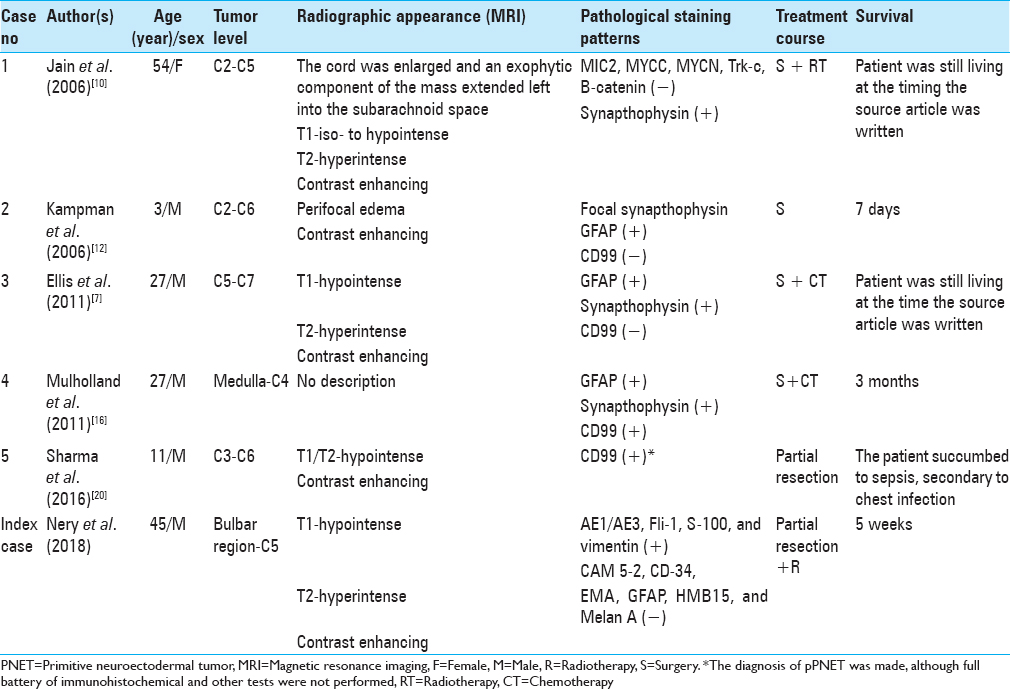
Background:Intramedullary primitive neuroectodermal tumors (PNETs) are tumors found rarely in the cervical region, with only five such cases described in the literature. The available literature contains only one report regarding cervicomedullary junction PNET.
Case Description:The authors present a case report of a 45-year-old male patient who had undergone urgent hospitalization owing to progressive tetraparesis and subtle impairment of respiratory function. He underwent magnetic resonance imaging, which showed an extensive enhancing cervical intramedullary tumor extending from C5 to the bulbar region. Since he developed severe impairment of respiratory function, he required tracheostomy. He then underwent microsurgery 2 days after his admission, and a partial tumor resection was performed. The pathological diagnosis of PNET of the cervicomedullary junction (CMJ) was made. He had slight worsening of strength after surgery with subsequent deterioration over the next 3 weeks. The tumor displayed aggressive growth; thus, radiotherapy was indicated. Unfortunately, he developed severe febrile neutropenia and died after 2 weeks of radiotherapy. Given the rarity of the condition, we wish to review the epidemiology, pathophysiology, and treatment options of his population.
Conclusion:Intramedullary PNETs of the cervical spine and CMJ are exceedingly rare in adults; treatment of such patients remains a challenge, despite the modern neurosurgical armamentarium that is available.
Keywords: Cervicomedullary, intramedullary, neuroectodermal tumor, pathology, primitive, treatment
INTRODUCTION
Primary tumors of the spinal cord represent only approximately 2%–4% of all primary tumors of the central nervous system (CNS); these spinal cord tumors can be divided into three anatomical categories: intramedullary, intradural extramedullary, and extradural.[
Spinal PNETs are further subclassified as a central subtype developing within the spinal cord and a peripheral subtype mainly occurring in the cauda equina.[
MATERIALS AND METHODS
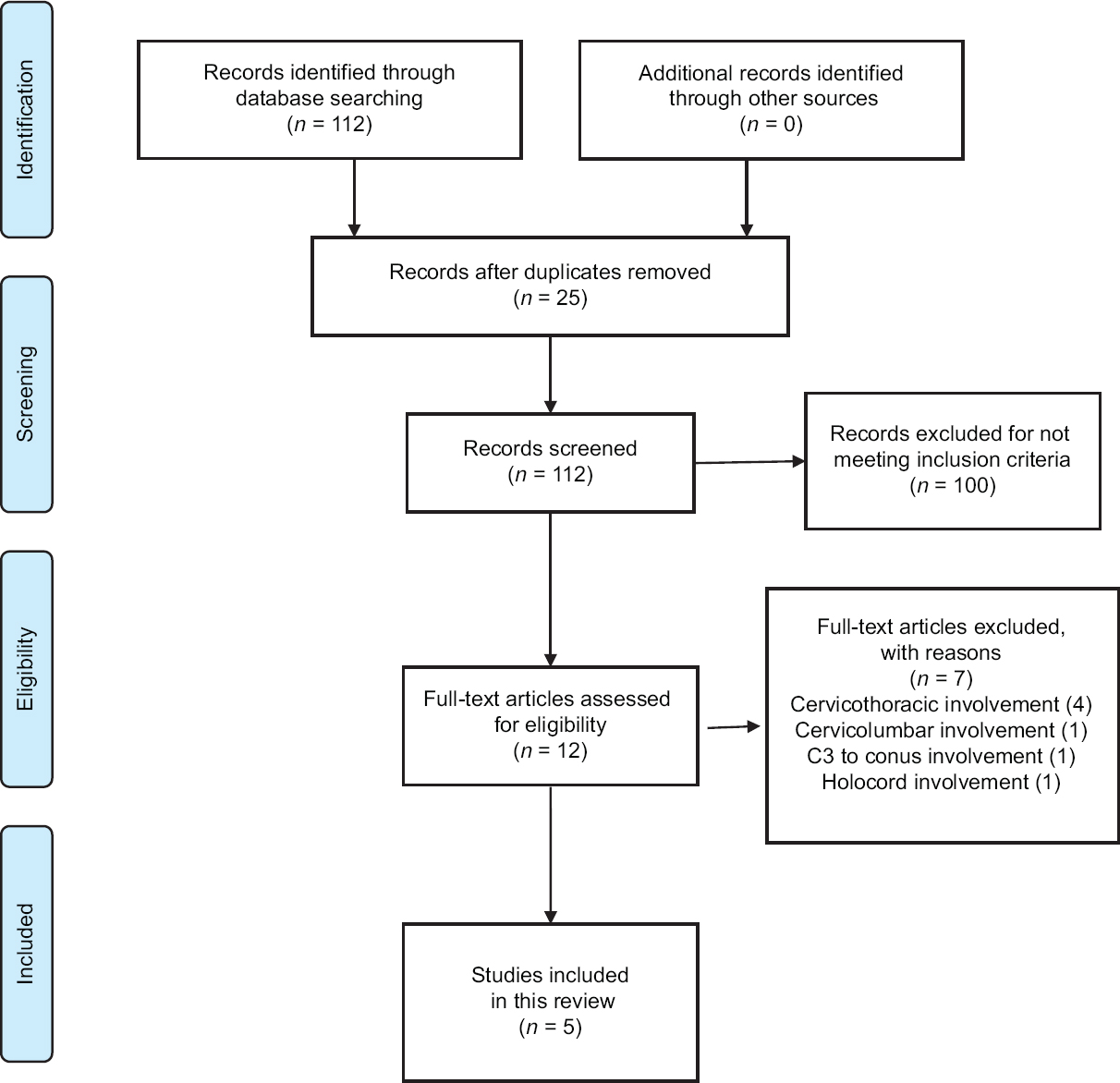
Through January 2018, a database search was performed through PubMed, ScienceDirect, EMBASE, and SciELO in order to identify eligible articles for the review. The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines were strictly followed throughout this review.
Search strategy
Studies were deemed eligible for inclusion if they report cervical intramedullary spinal PNET. The keywords used were “spinal cord neoplasms,” and “primitive neuroectodermal tumors.” The exclusion criteria included involvement of the thoracic, lumbar, or sacral levels.
RESULTS
A summary of the flow of studies through the review is presented in
CASE DESCRIPTION
In 2013, a 45-year-old male patient experienced progressive tetraparesis over about 1 week, which led him to seek medical attention. Based on the currently available British medical research council (MRC) grading system, neurosurgical evaluation revealed grade IV tetraparesis and global hyperreflexia with pyramidal liberation. Therefore, urgent brain and cervical spine magnetic resonance imaging (MRI) was requested, but he did not undergo said exam on the same day.
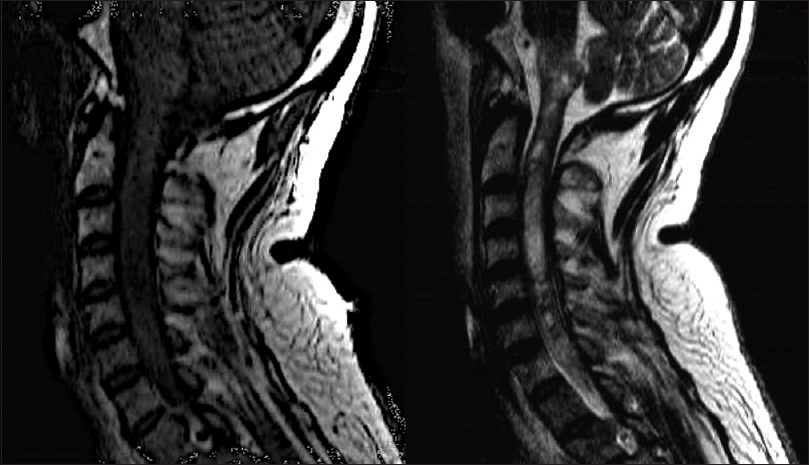
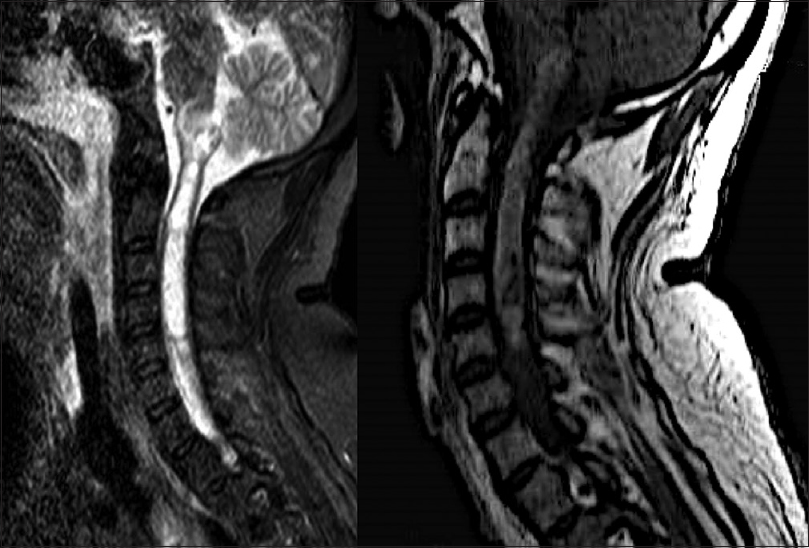
Four days after his first medical visit, he represented with subtle respiratory impairment, for which he was urgently admitted to our hospital for evaluation. An MRI was expeditiously obtained and showed an intramedullary lesion extending from the C5 level to the bulbar region with heterogeneous appearance and hypointense signal on T1-weigthed imaging, and without cystic or calcified components [
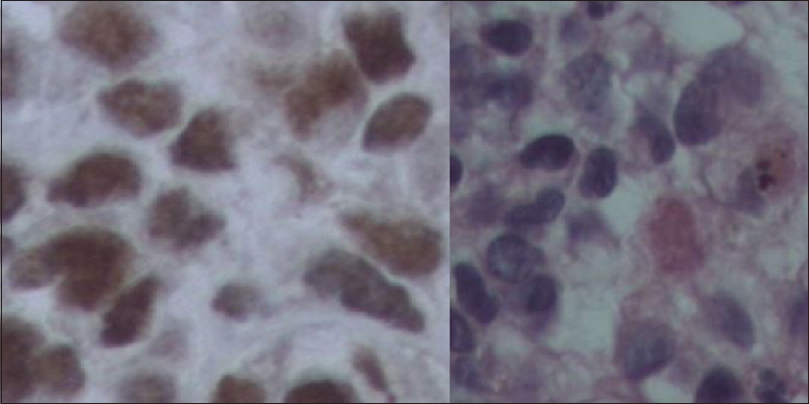
Immunohistochemical analysis revealed positive staining for the following markers: AE1/AE3, Fli-1, S-100, and vimentin. Besides this, CAM 5-2, CD-34, EMA, GFAP, HMB15, and Melan A stains were negative. Ten percent of the analyzed cells were positive for Ki-67, and the pathological characteristics were thought to be the most compatible with PNET.
Unfortunately, 3 weeks after establishing the diagnosis, he presented with severe worsening of motor and respiratory function, and with significant progression of the lesion on imaging, and he was subsequently referred for radiotherapy as adjuvant therapy. Due to clinical worsening, radiation of the neuroaxis was given only with 1,620 centigray. After 2 weeks of radiotherapy, he developed severe febrile neutropenia, and despite aggressive treatment, he died during the hospitalization.
DISCUSSION
Primary spinal cord tumors (PSCT) comprise 2%–4% of all primary nervous system tumors and are ~15 times less frequent than comparable primary intracranial tumors.[
Intramedullary tumors present with variable clinical manifestations such as pain and mixed sensorimotor or syringomyelic disturbances; furthermore, intramedullary tumors are often accompanied by dissociation of pain/temperature and proprioception. Purely intramedullary primary tumors are rare; PSCTs more commonly involve the thoracic and lumbar spine.
Here, we present a rare, purely intramedullary PNET in a unique location and this constitutes the second report of such a PNET involving the cervical spine and bulbar region.
Hart and Earle were responsible for creating the term “primary neuroectodermal tumors” in 1973 to describe predominantly undifferentiated brain tumors containing 90%–95% undifferentiated cells and not meeting diagnostic criteria for other tumor entities.[
PNETs were first defined as a new tumor entity in the World Health Organization Classification from 2007, which defines a PNET as an embryonic tumor composed of undifferentiated or poorly differentiated neuroepithelial cells that have the ability to exhibit a divergent differentiation along neuronal, ependymal, muscular, or melanocytic lines.[
Spinal PNETs occur most often in children[
PNETs can occur at any level of the spine, more commonly in the thoracic and lumbar spine.[

The immunohistopathological features of PNET include the following:
Small round or spindle cells with a low level of differentiation; Densely packed architecture or in the form of leaves or nests; Cells that are generally positive for neuronal markers such as neuron-specific enolase; they may also be positive for intermediate precursor filaments such as nestin, vimentin, or microfilaments; depending on cell differentiation, they may also be positive for synaptophysin or GFAP; it is also common for them to also be positive for S-100; In the case of undifferentiated cells, they cannot present positive results for any analysis.[
The diagnosis of a primary PNET of the spinal cord can be corroborated by MRI results, and exclusion of cranial involvement is necessary. PNET of the spinal cord is characterized by a rapid progression of the symptoms. When spinal cord biopsy results suggest the diagnosis of a primary PNET of the spinal cord, it is prudent to perform complete neuroaxial imaging, CSF cytology, and a whole-body PET scan. Due to their rarity, the diagnoses are generally not anticipated in the preoperative period.[
Complete surgical resection is considered the best treatment, which is usually followed by radiotherapy and chemotherapy. However, primary PNETs of the spinal cord are prone to metastasize throughout the subarachnoid space, so their prognosis is poor, and the tendency for metastases and relapses are common. Reported survival varied from several days to 36 months or alive at 15 months after diagnosis, with a median survival of more than a year.[
CONCLUSION
In conclusion, intramedullary PNETs are rare, particularly in the cervical spine and bulbar region. There remains a lack of information regarding the molecular pathology of such distinct PNETs and there are no established targeted treatment protocols available at present. Preoperative MRI study of the affected region and of the neuroaxis, as well as multiprofessional teamwork is paramount for treating PNETs. The rare occurrence of such lesions makes the presurgical diagnosis difficult, and despite possible complete resection of such lesions, treatment will probably have a poor outcome due to the location of the lesion, even when adjuvant radiotherapy and chemotherapy are administered. However, with increasing molecular data emerging allowing for targeted oncological therapy, we hope that with increasing knowledge about this distinct PNET type further treatment will emerge.
Authors’ contributions
BN, LCTP, RAFC, RMQ, LGA, EQ, LHC, ISTP, CHM, and FBF contributed to conception and design as well as the acquisition, analysis, and interpretation of data. RMQ, LGA, ISTP, and CHM contributed to acquisition and interpretation of imaging data. BN, LCTP, RAFC, RMQ, LGA, EQ, LHC, ISTP, CHM, and FBF documented the patient's status and contributed to analysis and interpretation of data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Albrecht CF, Weiss E, Schulz-Schaeffer WJ, Albrecht T, Fauser S, Wickboldt J. Primary intraspinal primitive neuroectodermal tumor: Report of two cases and review of the literature. J Neurooncol. 2003. 61: 113-20
2. Alexiou GA, Siozos G, Stefanaki K, Moschovi M, Prodromou N. Intramedullary spinal cord primitive neuroectodermal tumor presenting with hydrocephalus. J Child Neurol. 2013. 28: 246-50
3. Brat DJ, Parisi JE, Kleinschmidt-DeMasters BK, Yachnis AT, Montine TJ, Boyer PJ. Surgical neuropathology update: A review of changes introduced by the WHO classification of tumours of the central nervous system, 4th edition. Arch Pathol Lab Med. 2008. 132: 993-1007
4. Chamberlain MC, Tredway TL. Adult primary intradural spinal cord tumors: A review. Curr Neurol Neurosci Rep. 2011. 11: 320-8
5. Chen YC, Tang LM, Chen CJ, Jung SM, Chen ST. Intracranial hypertension as an initial manifestation of spinal neuroectodermal tumor. Clin Neurol Neurosurg. 2005. 107: 408-11
6. Coumans JV, Walcott BP, Nahed BV, Oh KS, Chi AS. Multimodal therapy of an intramedullary cervical primitive neuroectodermal tumor in an adult. J Clin Oncol. 2012. 30: e15-8
7. Ellis JA, Rothrock RJ, Moise G, McCormick PC, Tanji K, Canoll P. Primitive neuroectodermal tumors of the spine: A comprehensive review with illustrative clinical cases. Neurosurg Focus. 2011. 30: E1-
8. Elsberg CA. Some aspects of the diagnosis and surgical treatment of tumors of the spinal cord: With a study of the end results in a series of 119 operations. Ann Surg. 1925. 81: 1057-73
9. Fujisawa H, Kaneko T, Tohma Y, Kida S, Kaizaki Y. Central nervous system primitive neuroectodermal tumor of spinal cord developing 20 years after curative treatment of pineal tumor. Neurol Med Chir (Tokyo). 2011. 51: 596-9
10. Jain A, Jalali R, Nadkarni TD, Sharma S. Primary intramedullary primitive neuroectodermal tumor of the cervical spinal cord. Case report. J Neurosurg Spine. 2006. 4: 497-502
11. Jingyu C, Jinning S, Hui M, Hua F. Intraspinal primitive neuroectodermal tumors: Report of four cases and review of the literature. Neurol India. 2009. 57: 661-8
12. Kampman WA, Kros JM, De Jong TH, Lequin MH. Primitive neuroectodermal tumours (PNETs) located in the spinal canal; the relevance of classification as central or peripheral PNET: Case report of a primary spinal PNET occurrence with a critical literature review. J Neurooncol. 2006. 77: 65-72
13. Khmou M, Malihy A, Lamalmi N, Rouas L, Alhamany Z. Peripheral primitive neuroectodermal tumors of the spine: A case report and review of the literature. BMC Res Notes. 2016. 9: 438-
14. Kumar R, Reddy SJ, Wani AA, Pal L. Primary spinal primitive neuroectodermal tumor: Case series and review of the literature. Pediatr Neurosurg. 2007. 43: 1-6
15. Miller DC. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000. 47: 189-94
16. Mulholland CB, Barkhoudarian G, Cornford ME, McBride DQ. Intraspinal primitive neuroectodermal tumor in a man with neurofibromatosis type 1: Case report and review of the literature. Surg Neurol Int. 2011. 2: 155-
17. Ogasawara H, Kiya K, Kurisu K, Muttaqin Z, Uozumi T, Sugiyama K. Intracranial metastasis from a spinal cord primitive neuroectodermal tumor: Case report. Surg Neurol. 1992. 37: 307-12
18. Parsa AT, Chi JH, Acosta FL, Ames CP, McCormick PC. Intramedullary spinal cord tumors: Molecular insights and surgical innovation. Clin Neurosurg. 2005. 52: 76-84
19. Saeedinia S, Nouri M, Alimohammadi M, Moradi H, Amirjamshidi A. Primary spinal extradural Ewing's sarcoma (primitive neuroectodermal tumor): Report of a case and meta-analysis of the reported cases in the literature. Surg Neurol Int. 2012. 3: 55-
20. Sharma P, Das KK, Mehrotra A, Srivastava AK, Sahu RN, Jaiswal A. Cervicomedullary intramedullary peripheral primitive neuroectodermal tumor with intratumoral bleed: Report of one case and review of literature. J Craniovertebr Junction Spine. 2016. 7: 111-4
21. Smoll NR, Drummond KJ. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci. 2012. 19: 1541-4
22. Wang G, Guo F. Primary intramedullary primitive neuroectodermal tumor: A case report and review of the literature. Medicine (Baltimore). 2017. 96: e9001-

