

- Department of Neurological Surgery, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA
- Department of Pathology, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA
- Department of Neurology, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA
Correspondence Address:
Daniel A. Tonetti
Department of Neurology, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA
DOI:10.4103/2152-7806.210257
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Daniel A. Tonetti, William J. Ares, R. Mark Richardson, Ronald L. Hamilton, Frank S. Lieberman. Long-term recurrence of dysembryoplastic neuroepithelial tumor: Clinical case report. 11-Jul-2017;8:140
How to cite this URL: Daniel A. Tonetti, William J. Ares, R. Mark Richardson, Ronald L. Hamilton, Frank S. Lieberman. Long-term recurrence of dysembryoplastic neuroepithelial tumor: Clinical case report. 11-Jul-2017;8:140. Available from: http://surgicalneurologyint.com/surgicalint-articles/long%e2%80%91term-recurrence-of-dysembryoplastic-neuroepithelial-tumor-clinical-case-report/
Date of Submission
18-Oct-2016
Date of Acceptance
10-Nov-2016
Date of Web Publication
11-Jul-2017
Abstract
Background:Dysembryoplastic neuroepithelial tumors (DNETs) are rare, benign brain neoplasms that typically arise in children and adolescents and classically present with intractable, partial complex seizures. DNETs are classically associated with a favorable prognosis after complete surgical resection.
Case Description:We describe a case of long-term recurrence of a DNET, which initially resected and diagnosed as an oligodendroglioma prior to the recognition of DNETs. This patient was seizure-free for 12 years and had no signs of radiologic progression until 24 years after initial resection. On repeat surgical resection, 31 years after the initial surgery, histopathologic evaluation identified the characteristic features of DNET in both specimens.
Conclusions:This patient's 24-year disease-free interval prior to radiologic recurrence demonstrates the longest interval to relapse in the literature for a DNET. This case illustrates the possibility of late recurrence of DNETs decades after radiographical complete resection to emphasize the necessity of thoughtful clinical judgment in adult survivors of low grade pediatric neoplasms who present with seizures after a prolonged seizure-free interval.
Keywords: Dysembryoplastic neuroepithelial tumor, low-grade glioma, recurrence
INTRODUCTION
First described in 1988,[
DNETs are slow-growing benign neoplasms associated with a favorable prognosis after complete resection without subsequent radiation or adjuvant chemotherapy. In contrast to subtotal resection, gross total resection of DNETs appears to correlate with low recurrence rates and improved seizure control,[
CASE REPORT
In 1982, a 9-year-old right-handed Caucasian boy with no past neurologic history experienced a first-time grand-mal seizure and underwent neuroimaging, which demonstrated a mass lesion involving the right posterior parieto-occipital region. He subsequently underwent a reported gross surgical resection, which led to a diagnosis of low-grade oligodendroglioma. Postoperatively, the patient remained seizure-free for 12 years until age 21, when he began experiencing both simple and complex partial seizures 1–2 times monthly. He was placed on carbamazepine and gabapentin, and an MRI at that time was unrevealing for recurrent tumor.
From age 21 to 31, the patient underwent serial MRI scans every other year, each of which demonstrated a stable resection cavity with no disease recurrence when compared to prior examinations. In 2006, at age 33, the patient's surveillance MRI demonstrated a nonenhancing mass spanning the right parieto-occipital junction without peritumoral edema or mass effect, concerning for tumor recurrence in the resection cavity of the surgery 24 years prior. The patient elected to defer surgery and continue medical management of his epilepsy. Over the next several years, he continued to develop worsening seizures refractory to three antiepileptic medications. Surveillance MRIs demonstrated progressive nodular enhancement of the lesion [
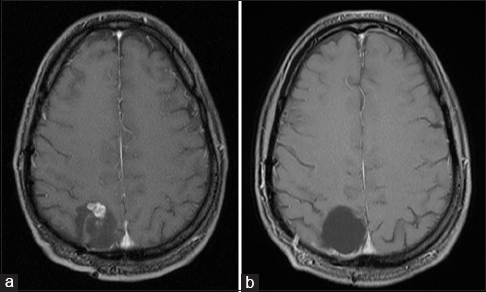
Histopathological evaluation identified the characteristic features of DNET in both specimens [
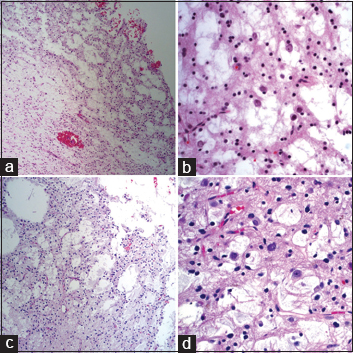
Figure 2
Hematoxylin and eosin (H and E) stained sections from 1982 at ×100 (a) show a round-cell glial lesion with microcystic features and myxoid change consistent with an oligodendroglioma. At ×400 (b), there are neurons floating in mucinous pools, a distinctive feature of dysembryoplastic neuroepithelial tumor (DNET) that was not identified until 1988. H and E stained sections from 2013 show almost identical features, now widely recognized as characterizing a DNET (c, ×100; d, ×400)
DISCUSSION
Since the initial study of a recurrent case in 2000,[
Seizure recurrence after gross total resection for DNET is not uncommon, and our patient developed seizures 12 years after his initial resection. Although complete resection of the tumor is sufficient to cure epilepsy related to DNET in many cases,[
Radiologic features of DNETs include hypointensity on T1-weighted MR images and hyperintensities on T2-weighted MR images whereas edema and mass effect are commonly absent.[
This case highlights the necessity of thoughtful clinical judgment in patients who present with epilepsy after a prolonged seizure-free interval. DNET should be considered in the differential diagnosis for patients with long-standing epilepsy and tumor recurrence, particularly if they underwent surgical resections of neoplasms prior to the recognition of DNETs in 1988.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Bilginer B, Yalnizoglu D, Soylemezoglu F, Turanli G, Cila A, Topçu M. Surgery for epilepsy in children with dysembryoplastic neuroepithelial tumor: Clinical spectrum, seizure outcome, neuroradiology, and pathology. Childs Nerv Syst. 2009. 25: 485-91
2. Chao L, Tao XB, Jun YK, Xia HH, Wan WK, Tao QS. Recurrence and histological evolution of dysembryoplastic neuroepithelial tumor: A case report and review of the literature. Oncol Lett. 2013. 6: 907-14
3. Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Vedrenne C. Dysembryoplastic neuroepithelial tumor: A surgically curable tumor of young patients with intractable partial seizures. Report of 39 cases. Neurosurgery. 1988. 23: 545-56
4. Daumas-Duport C, Varlet P, Bacha S, Beuvon F, Cervera-Pierot , Chodkiewicz JP. Dysembryoplastic neuroepithelial tumors: Nonspecific histological forms – A study of 40 cases. J Neurooncol. 1999. 41: 267-80
5. Englot DJ, Berger MS, Barbaro NM, Chang EF. Factors associated with seizure freedom in the surgical resection of glioneuronal tumors. Epilepsia. 2012. 53: 51-7
6. Fernandez C, Girard N, Paredes AP, Bouvier-Labit C, Lena G, Figarella-Branger D. The usefulness of MR imaging in the diagnosis of dysembryoplastic neuroepithelial tumor in children: A study of 14 cases. AJNR Am J Neuroradiol. 2003. 24: 829-34
7. Hammond RR, Duggal N, Woulfe JM, Girvin JP. Malignant transformation of a dysembryoplastic neuroepithelial tumor. Case report. J Neurosurg. 2000. 92: 722-5
8. Jensen RL, Caamano E, Jensen EM, Couldwell WT. Development of contrast enhancement after long-term observation of a dysembryoplastic neuroepithelial tumor. J Neurooncol. 2006. 78: 59-62
9. Khajavi K, Comair YH, Wyllie E, Palmer J, Morris HH, Hahn JF. Surgical Management of pediatric tumor-associated epilepsy. J Child Neurol. 1999. 14: 15-25
10. Kim AH, Thompson EA, Governale LS, Santa C, Cahill K, Kieran MW. Recurrence after gross-total resection of low-grade pediatric brain tumors: The frequency and timing of postoperative imaging. J Neurosurg Pediatr. 2014. 14: 356-64
11. Kuroiwa T, Bergey GK, Rothman MI, Zoarski GH, Wolf A, Zagardo MT. Radiologic appearance of the dysembryoplastic neuroepithelial tumor. Radiology. 1995. 197: 233-8
12. Luyken C, Blümcke I, Fimmers R, Urbach H, Elger CE, Wiestler OD. The spectrum of long-term epilepsy-associated tumors: Long-term seizure and tumor outcome and neurosurgical aspects. Epilepsia. 2003. 44: 822-30
13. Maher CO, White JB, Scheithauer BW, Raffel C. Recurrence of dysembryoplastic Neuroepithelial tumor following resection. Pediatr Neurosurg. 2008. 44: 333-6
14. Nolan MA, Sakuta R, Chuang N, Otsubo H, Rutka JT, Snead OC III. Dysembrytoplastic neuroepithelial tumors in childhood: Long-term outcome and prognostic features. Neurology. 2004. 62: 2270-6
15. Paudel K, Borofsky S, Jones RV, Levy LM. Dysembryoplastic neuroepithelial tumor atypical presentation: MRI and diffusion tensor characteristics. J Radiol Case Rep. 2013. 7: 7-14
16. Santos MV, de Oliveira RS, Machado HR. Approach to cortical dysplasia associated with glial and glioneuronal tumors (FCD type IIIb). Childs Nerv Syst. 2014. 30: 1869-74
17. Yu AH, Chen L, Li YJ, Zhang GJ, Li KC, Wang YP. Dysembryoplastic neuroepithelial tumors: Magnetic resonance imaging and magnetic resonance spectroscopy evaluation. Chin Med J. 2009. 122: 2433-7

