

- Department of Neurological Surgery, University of Kansas Medical Center, Kansas City, Kansas, USA
- Department of Neuro-Oncology, University of Kansas Medical Center, Kansas City, Kansas, USA
Correspondence Address:
Anthony M. Alvarado
Department of Neurological Surgery, University of Kansas Medical Center, Kansas City, Kansas, USA
DOI:10.4103/sni.sni_24_17
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Anthony M. Alvarado, Michael E. Salacz, Roukoz B. Chamoun. Malignant glioma-primitive neuroectodermal tumor recurring as PNET-like only subdural collection: Case report. 10-Oct-2017;8:243
How to cite this URL: Anthony M. Alvarado, Michael E. Salacz, Roukoz B. Chamoun. Malignant glioma-primitive neuroectodermal tumor recurring as PNET-like only subdural collection: Case report. 10-Oct-2017;8:243. Available from: http://surgicalneurologyint.com/surgicalint-articles/malignant-glioma%e2%80%91primitive-neuroectodermal-tumor-recurring-as-pnet%e2%80%91like-only-subdural-collection-case-report/
Date of Submission
22-Jan-2017
Date of Acceptance
04-Aug-2017
Date of Web Publication
10-Oct-2017
Abstract
Background:Histologic variants of conventional glioblastoma are rare clinical entities. In recent years, an aggressive variant termed malignant glioma with primitive neuroectodermal tumor components (MG-PNET) has been described in adults. In addition to the rarity of supratentorial primitive neuroectdoermal tumors (sPNET) in adults, MG-PNET can present with unique radiographic features.
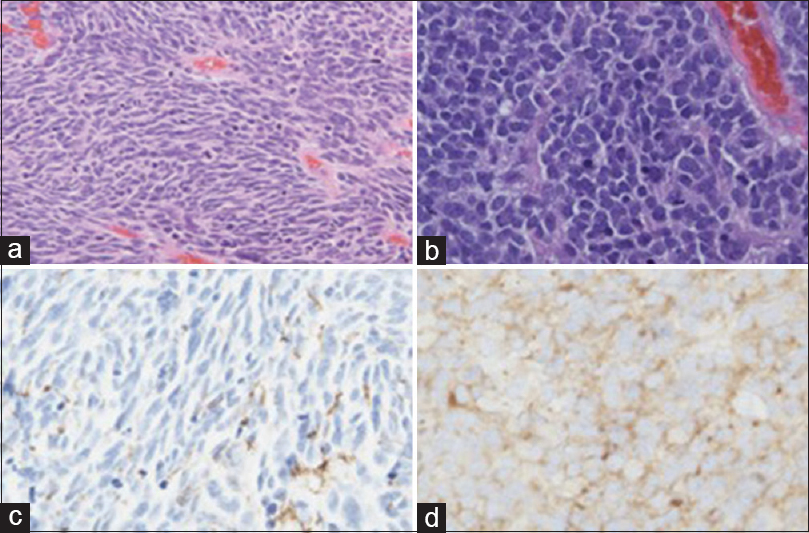
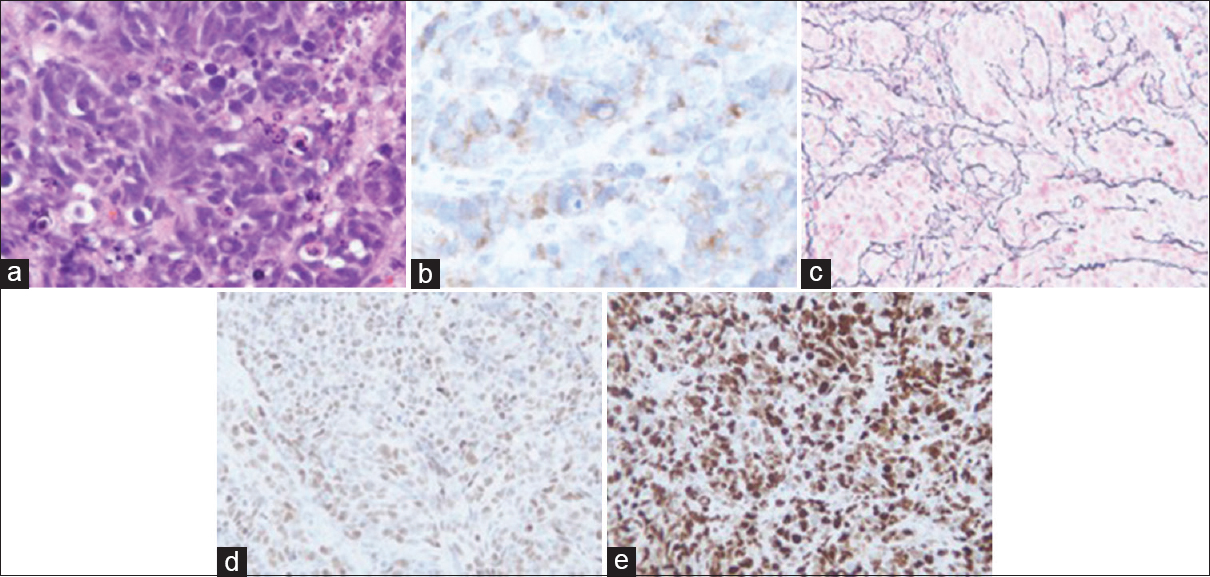
Case Description:We report the case of a 42-year-old male who presented with headaches and vision changes. Magnetic resonance imaging (MRI) of the brain revealed a large right frontal lesion. He underwent craniotomy with pathology demonstrating glioblastoma WHO grade IV, with primitive neuroectodermal tumor-like components (MG-PNET). Seven weeks later the patient represented with worsening headaches and left-hand weakness. MRI brain revealed a diffusion restricting subdural collection overlying the prior craniotomy site. Biopsy revealed PNET-like recurrence of the previously treated MG-PNET.
Conclusion:In addition to histologic deviation, MG-PNET can present with variable radiographic findings on MRI and a clinical course distinctive from traditional glioblastoma. The hypercellular nature of this lesion can present as a diffusion-restricting lesion.
Keywords: Diffusion-weighted MRI, glioblastoma, platinum-based chemotherapy, primitive neuroectodermal tumor, temozolomide
INTRODUCTION
Glioblastoma is an aggressive, high-grade glioma and is the most common primary malignant brain tumor in adults.[
Clinical behavior and treatment approach vary with the individual components of MG-PNET. Glioblastomas are glial neoplasms, staining briskly with glial fibrillary acidic protein (GFAP) and most often present in adulthood. Treatment consist of surgical resection followed by radiation therapy and chemotherapy with alkylating agents such as temozolomide.[
In comparison, supratentorial PNETs (distinct from medulloblastoma) are predominately neuronal tumors, appearing as small round blue-cell tumors and staining for synaptophysin and neuron-specific enolase (NSE). These tumors primarily affect children, retain a high proliferation index, and have the potential for cerebrospinal fluid (CSF) dissemination.[
MG-PNET is difficult to diagnose radiographically due to their rarity, microscopic (as opposed to macroscopic) areas of neuronal tumor within the more dominant glial tumor, and lack of large studies. Prior work has reported the use of diffusion-weighted MRI in diagnosing suspected lesions as they have the potential to demonstrate reduced apparent diffusion coefficient (ADC) values in areas containing hypercellular foci, compared to conventional GBM. Here, we report a case of a MG-PNET, initially resected, recurring as PNET-only histology and presenting as diffusion restricted subdural collection on MRI.
CASE REPORT
A 40-year-old male presented with 2-week history of subjective headache, nausea, and blurry vision. MRI brain [
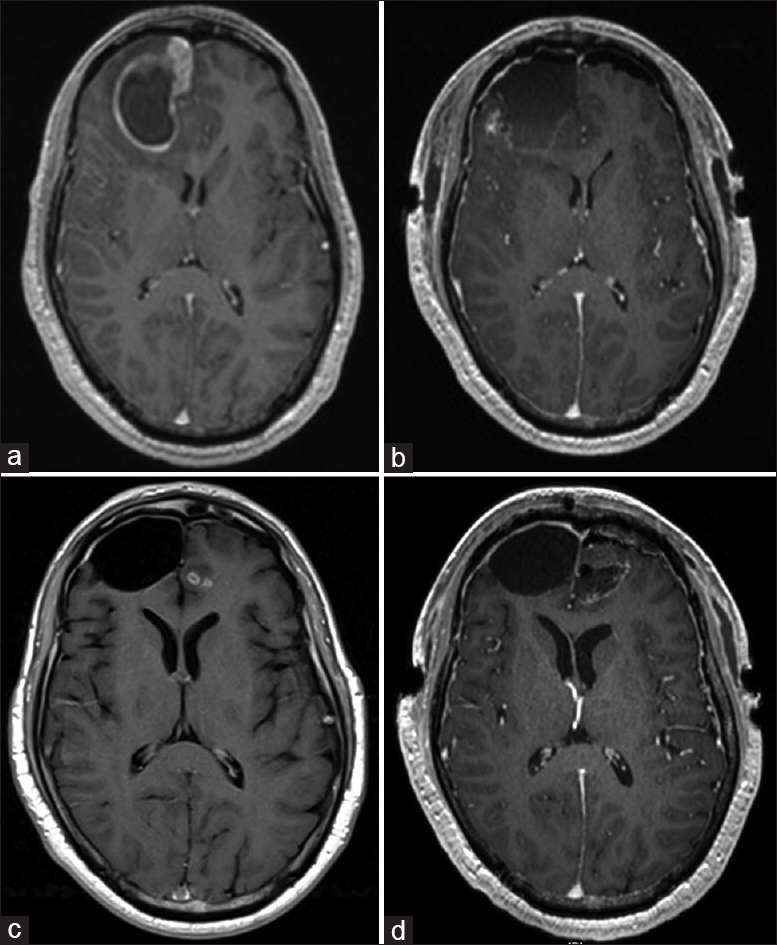
Figure 1
(a) MRI brain with contrast demonstrated 4.7 cm multiloculated rim enhancing cystic lesion with additional smaller projections invading the right frontal lobe with associated vasogenic edema and midline shift. (b) MRI brain with contrast demonstrating resection of right frontal mass with expected postoperative changes. (c) MRI brain with contrast demonstrating enhancing foci within left frontal lobe adjacent to prior surgical cavity. (d) MRI brain with contrast demonstrating resection of left frontal foci with expected postoperative changes
Due to the PNET-like component of the tumor, MRI of the spinal axis was performed and was without evidence of drop metastasis. Subsequent treatment with radiation therapy and concurrent temozolomide chemotherapy was completed. Twelve weeks after the initial resection and post radiation therapy/temozolomide, surveillance MRI scan revealed multiple new enhancing foci within the left frontal lobe [
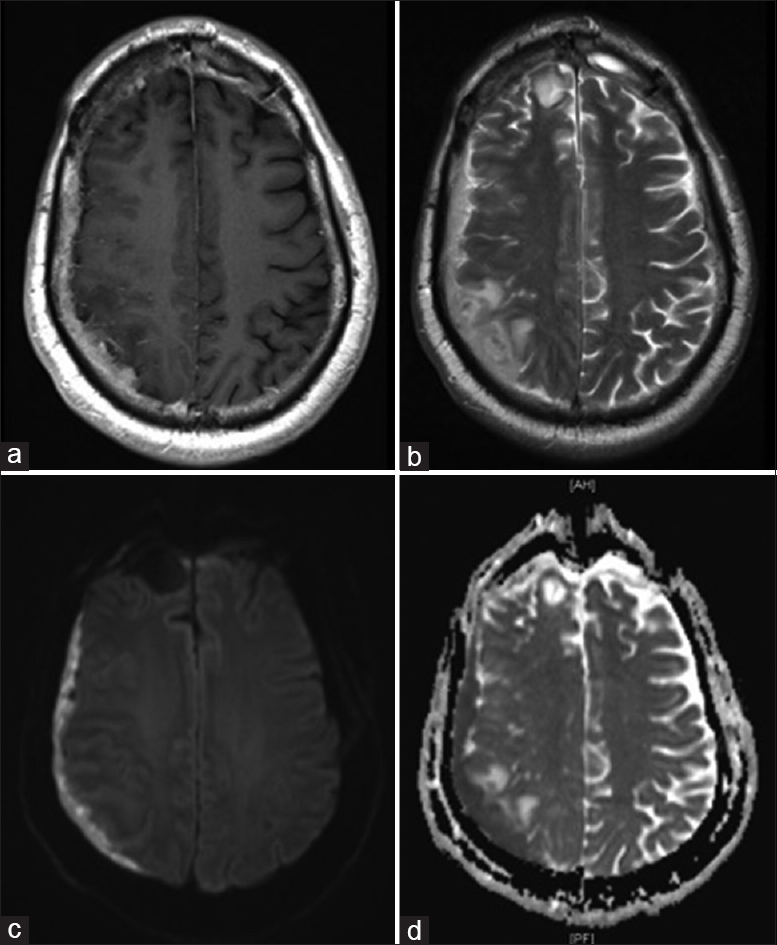
Because PNET is typically treated with platinum-based chemotherapy, studies confirmed MGMT unmethylated status and demonstrated potential sensitivity to platinum-based chemotherapy. In addition, biomarker results demonstrated topoisomerase activity. Given these findings and that PNET is traditionally treated with platinum-based chemotherapy, the patient was treated with salvage cisplatin and irinotecan. These agents were selected as cisplatin gains superior central nervous system penetration and irinotecan has been used in this setting on primary brain tumors. Given the toxicity associated with the selected regimen, a long discussion was held with the patient prior to initiating therapy. Initially, the patient responded well both clinically and radiographically to therapy; however, surveillance imaging at 8 weeks demonstrated significant tumor progression [
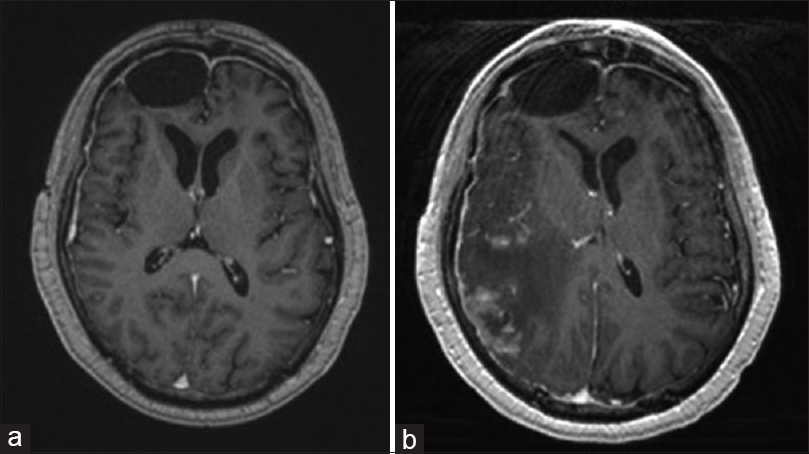
Figure 5
(a) MRI T1W with contrast four-weeks post-surgical evacuation of subdural collection and initiation of platinum-based chemotherapy demonstrating marked improvement. (b) MRI T1W with contrast eight-weeks following initiation platinum-based chemotherapy demonstrating marked progression of invasive tumor in the right parietal and posterior frontal lobes, extensive vasogenic edema extending to the atrium of the right lateral ventricle and midline shift
DISCUSSION
MG-PNET represents a rare histological variant of high-grade glioma. The reported overall frequency of PNET-like components appearing with glioblastoma has been estimated in 1 out of 200 cases.[
Neurologic dysfunction in GBM-PNET is related to rapid tumor growth, peritumoral edema, and elevated intracranial pressure. Manifestations include headaches, nausea, vomiting, and seizures. Furthermore, focal neurological deficits including visual field defects, aphasia, extremity paresis, and facial nerve palsy may present depending on the anatomic location of the neoplasm.[
Even so, the genetic features and prognosis of adult PNET are still widely uncertain. It is known that pediatric PNETs associated with c-myc and N-myc gene amplifications are associated with a decreased survival. A review by Gessi et al. on supratentorial PNET occurrence in adults did not demonstrate amplification of c-myc/N-myc genes, thus indicating that PNET in adults may represent a specific subset of tumors.[
The clinical features and behavior of GBM-PNET are variable, and the knowledge regarding its associated molecular markers is largely unknown. In a review of 53 patients of MG-PNET, Perry et al. concluded that the median age of diagnosis was 54 years ranging 21–80 years.[
Aside from the difficulties in treating GBM-PNET, radiographic presentation varies considerably and imaging characteristics are not fully understood. Though there reports of PNET presenting as intracranial hemorrhage (ICH) are rare,[
CONCLUSION
The imaging characteristics of GBM-PNET are not well described and often propose a diagnostic dilemma. In the setting of glioblastoma neoplasms demonstrating restricted diffusion and lower ADC values compared to conventional glioblastoma, clinical suspicion for PNET-like components should remain elevated as areas of restriction may represent hypercellular neoplastic tissue. Moreover, DWI sequence utilization can assist in obtaining tissue for accurate histopathological diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ali S, Joseph NM, Perry A, Barajas RF, Cha S. Apparent diffusion coefficient in glioblastoma with PNET-like components, a GBM variant. J Neurooncol. 2014. 119: 353-60
2. Calli C, Kitis O, Yunten N, Yurtseven T, Islekel S, Akalin T. Perfusion and diffusion MR imaging in enhancing malignant cerebral tumors. Eur J Radiol. 2006. 58: 394-403
3. Dresemann G. Temozolamide in malignant glioma. Onco Targets Ther. 2010. 3: 139-40
4. Forbes V, Vredenburgh J. Primitive Neuroectodermal Tumor with Glioblastoma Multiforme Components in an Adult: A Collision Tumor. Cureus. 2016. 8: e456-
5. Gessi M, Setty P, Bisceglia M, Muehlen A, Lauriola L, Waha A. Supratentorial Primitive Neuroectodermal Tumors of the Central Nervous System in Adults: Molecular and Histopathologic Analysis of 12 Cases. Am J Surg Pathol. 2011. 35: 573-82
6. Ishizawa K, Kan-nuki S, Kumagai H, Komori T, Hirose T. Lipomatous primitive neuroectodermal tumor with a glioblastoma component: A case report. Acta Neuropathol. 2002. 103: 193-8
7. Kim DG, Lee DY, Paek SH, Chi JG, Choe G, Jung HW. Supratentorial primitive neuroectodermal tumors in adults. J Neurooncol. 2002. 60: 43-52
8. Papadopoulos EK, Fountas KN, Brotis AG, Paterakis KN. A supratentorial primitive neuroectodermal tumor presenting with intracranial hemorrhage in a 42-year-old man: A case report and review of the literature. J Med Case Rep. 2013. 7: 86-
9. Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC. Malignant Gliomas with Primitive Neuroectodermal Tumor-like Components; A Cliniopathologic and Genetic Study of 53 cases. Brain Pathol. 2009. 19: 81-90
10. Song X, Andrew Allen R, Terence Dunn S, Fung KM, Farmer P, Gandhi S. Glioblastoma with PNET-like components has a higher frequency of isocitrate dehydrogenase 1 (IDH1) mutation and likely a better prognosis than primary glioblastoma. Int J Clin Exp Pathol. 2011. 4: 651-60
11. Stupp R, Mason WP, Van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. Radiotherapy plus concomitant and adjuvant temozolamide for glioblastoma. N Engl J Med. 2005. 352: 987-96

