

- Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Texas, USA
- Department of Neurobiology, Drexel University College of Medicine, Philadelphia, USA
Correspondence Address:
Sandi Lam
Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Texas, USA
DOI:10.4103/2152-7806.196922
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Visish M. Srinivasan, Michael G. Z. Ghali, Robert Y. North, Zain Boghani, Daniel Hansen, Sandi Lam. Modern management of medulloblastoma: Molecular classification, outcomes, and the role of surgery. 28-Dec-2016;7:
How to cite this URL: Visish M. Srinivasan, Michael G. Z. Ghali, Robert Y. North, Zain Boghani, Daniel Hansen, Sandi Lam. Modern management of medulloblastoma: Molecular classification, outcomes, and the role of surgery. 28-Dec-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/modern-management-of-medulloblastoma-molecular-classification-outcomes-and-the-role-of-surgery/
Date of Submission
20-Aug-2016
Date of Acceptance
14-Oct-2016
Date of Web Publication
28-Dec-2016
Keywords: Medulloblastoma, pediatric, posterior fossa syndrome
ILLUSTRATIVE CASE
A 4-year-old boy presented with papilledema and nystagmus. He had developed eye deviation, clumsiness, and episodes of dizziness starting 6 months prior to presentation, which progressed in frequency. Following an episode of emesis, he was brought to our attention. One week prior to presentation, the patient had one episode of emesis in the morning. His development had been notable for speech delay, with expressive language limited to speaking in short phrases without full sentence formation; he otherwise had normal growth and motor development.
Physical examination was significant for sluggish and dilated pupils, agitation, crying, and attention difficulty. Computer tomography (CT) scan of the brain revealed a large posterior fossa mass arising from the vermis with multiple calcifications and associated obstructive hydrocephalus. Magnetic resonance imaging (MRI) with and without contrast of the brain showed a mass with patchy enhancement and associated metastatic lesions located throughout the cerebellar hemispheres [
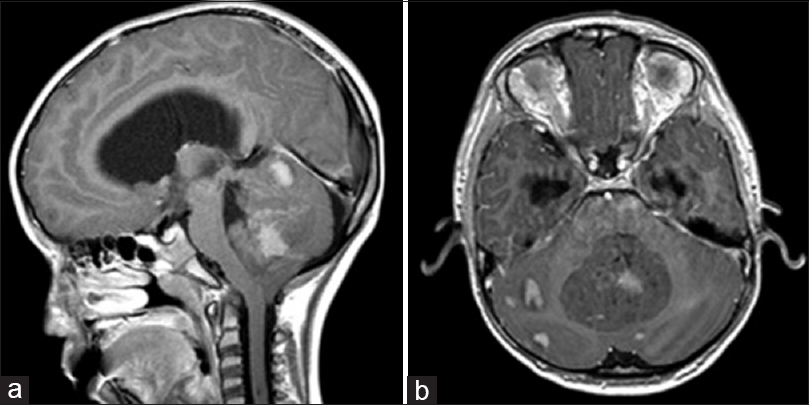
Figure 1
Preoperative MRI Brain, T1 post-gadolinium in sagittal (a) and axial (b) sections from patient in the example case. The large midline medulloblastoma arises from the cerebellar vermis and compresses the fourth ventricle, causing obstructive hydrocephalus. There is nodular leptomeningeal spread throughout the posterior fossa
Once in the operating room, a frontal external ventricular drain was placed prior to positioning the patient prone for a midline suboccipital craniotomy with transvermian approach splitting the inferior aspect of the vermis. The tumor was highly vascular and noted to be involving the floor of the fourth ventricle, bilateral foramina of Luschka, and left cerebellar peduncle, precluding a complete resection. Histopathology confirmed a medulloblastoma with subsequent molecular definition of a non-WNT(wingless)/non-SHH(sonic hedgehog) subgroup.
Postoperatively, the patient exhibited decreased responsiveness, mutism, fixed downward gaze, and inability to follow commands. Postoperative imaging did not demonstrate any hemorrhage [
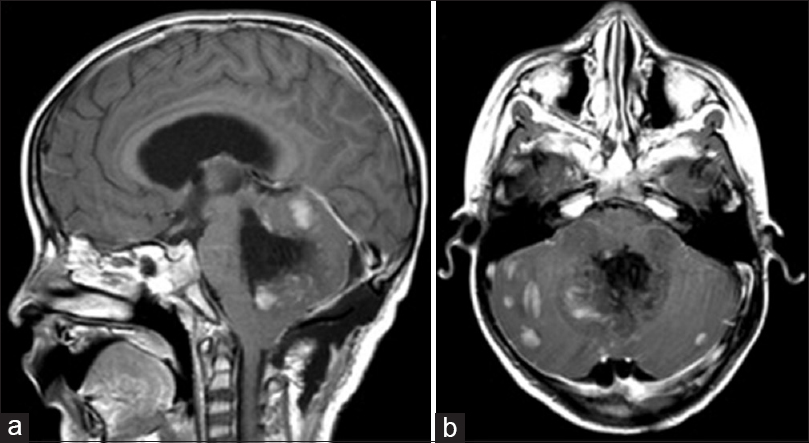
OVERVIEW OF MEDULLOBLASTOMA
Medulloblastoma (MB) is the most common posterior fossa tumor in children, presenting at a mean age of 9 years and, more commonly, in males (ratio 2:1).[
Molecular subgrouping
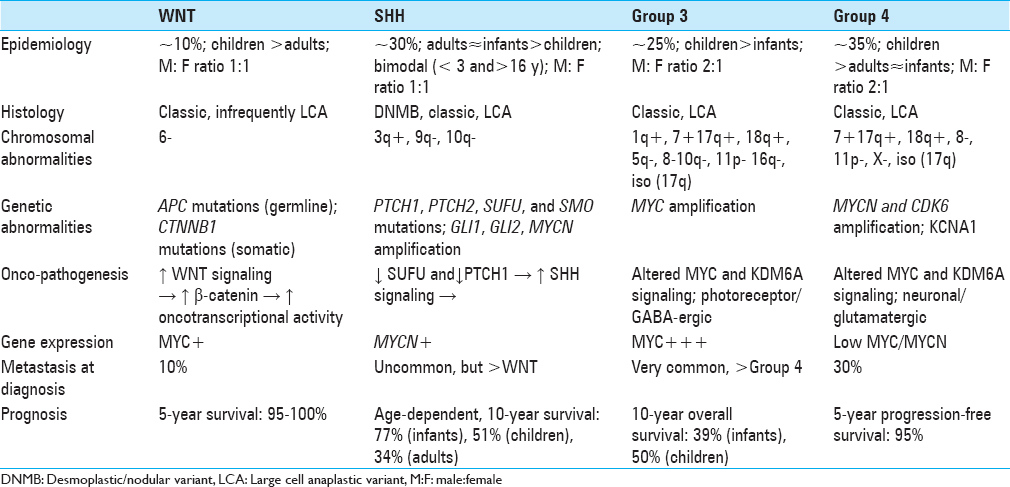
In addition to traditional histologic classification [classic, desmoplastic/nodular (DNMB), MB with extensive nodularity (MBEN), large cell/anaplastic (LCA)), MB has been classified into four distinct molecular subgroups according to transcriptional profiling studies.[
Genetics
The WNT and SHH classifications identify the underlying oncopathogenic pathway, whereas Groups 3 and 4 retain generic designations pending elucidation of underlying cellular pathobiology. WNT tumors result from unregulated WNT signaling leading to increased beta-catenin-mediated increase in transcriptional activity and consequent tumorigenesis. They are associated with monosomy 6, germline APC mutations (Turcot syndrome), somatic CTNNB1 mutations, and nuclear positivity for β-catenin.[
The precise oncopathogenic mechanisms for non-WNT/SHH tumors are still under investigation. Altered MYC and KDM6A signaling has been implicated in Groups 3 and 4 MBs. Whereas MYC is often overamplified in Group 3 MBs, Group 4 MBs are occasionally characterized by MYCN and CDK6 amplification. Isochromosome 17q is characteristic of Group 4 tumors but is also occasionally observed in Group 3 MBs, making it poorly specific. An alternative proposed marker for Group 4 lesions is KCNA1.[
Whereas molecular subgrouping of MB is based on differential gene transcriptional profiles, the different subgroups are recapitulated by subgroup-specific differential heterogeneity of cis-regulatory elements in the epigenome.[
Another compelling area of investigation in the genetics of MB is recurrent disease. Much of our understanding of the molecular and genetic profiles of MB is derived from experiments with treatment naïve and primary site disease. However, much like other malignancies, there is mounting evidence that suggests clonal selection and genetic divergence may play an important role in MB recurrence.[
Histologic correlation
There is an association between molecular subgroup and histologic type. For instance, 97% of WNT MBs are of the classic histologic variant.[
Epidemiologic correlation
WNT, SHH, Group 3, and Group 4 MBs account for 10%, 30%, 25%, and 35% of MBs overall, respectively, with a 1:1 M:F ratio for WNT and SHH subgroups, and a 2:1 male predominance for non-SHH/WNT tumors. WNT tumors are typically seen in children and adults, whereas Group 3 tumors are more often seen in infants and children. SHH and Group 4 tumors are seen across all age groups, with the former exhibiting a bimodal age distribution, most typically occurring in patients <4 and >16 years of age.[
Surgical treatment
Extent of resection
The extent of resection indicated in MBs largely depends on the unique anatomy of the tumor and what can be done safely and without the incurrence of neurological deficit, as with all tumors in the eloquent areas of brain. Although no clinical trials have been designed to specifically evaluate the role of surgery for MB, there have been many studies supporting a relationship of extent of resection with event-free survival. The most influential is likely a retrospective analysis of 233 children involved in a randomized controlled trial of differing chemotherapy regimens by Albright et al. determining that a radiographically measured residual tumor less than 1.5 cm3 was associated with improvement in 5-year PFS of greater than 20% in patients with M0 disease and an 11% difference for all patients irrespective of age, M stage, or any other measured factors.[
Hydrocephalus
Following resection, between 10 and 40% of the patients have hydrocephalus requiring CSF diversion.[
Adjuvant therapies
Risk stratification and radiotherapy
Traditionally, children older than 3 years of age are stratified into average and high-risk prognostic groups based on the presence of metastatic disease and post-resection residual less or greater than 1.5 cm3. “High-risk” MB is defined as having any one of the following characteristics: >1.5 cm3 postoperative residual, evidence of radiographic metastases, or presence of leptomeningeal disease/CSF seeding, with the remaining patients defined as “average-risk.”[
Under the above scheme, “high-risk” patients undergo posterior fossa or surgical bed radiation (54–55.8 Gy) with high-dose CSI (36.0 Gy) followed by adjuvant chemotherapy, whereas “average-risk” patients undergo posterior fossa or surgical bed radiation (54–55.8 Gy) with reduced-dose CSI (23.4 Gy) followed by adjuvant chemotherapy.[
Chemotherapy
Cytotoxic chemotherapy may be used in the initial treatment, maintenance therapy, or for recurrent disease. It may be radiation sparing, which would allow one to eschew the use of radiation in children <3 years of age and permit dose-reduction in older patients. Various regimens exist for initial treatment, with standard therapy being post-radiation cisplatin-based chemotherapy for 4–9 cycles.[
Many previous trials investigating chemotherapy and radiation regimens have compared outcomes based on the histologic type. However, the histologic types and molecular subgroups have not been completely congruent. Following the new stratification of MB by molecular subgroup, inroads have been made to tailor therapies to these pathways or predict response to traditional therapy.[
Specifically targeted chemotherapies based on pathways believed to be involved in oncogenesis are a promising future application of molecular subgrouping. At present, molecularly targeted agents for each of the four molecular subgroups are being studied in either clinical and pre-clinical models—the most well-studied of these being smoothened (SMO) receptor antagonists, such as vismodigib, that have demonstrated some utility in both preclinical and clinical models.[
Alternative strategies include sensitization of MB tumor cells to chemotherapeutic treatment. For example, thiostrepton, an antagonist of FOXM1 (an oncogene shown to be upregulated in a variety of malignancies), was shown to sensitize MB cells to cisplatin in vitro.[
Options for recurrent disease are more limited. A regimen of ifosfamide, cisplatin, and etoposide has been investigated but may be limited by significant attendant toxicities, most notable for profound myelosuppression.[
Prognosis and outcomes
The overall prognosis of MB is relatively good compared to other high-grade tumors, with a 5-year overall survival of approximately 70%.[
Histological phenotype has classically been very important in prognostication, especially in young children in whom it is highly predictive of outcome. Favorable results can be seen in DNMBs, which account for half of MB cases seen in children ≤3 years old.[
The WNT subgroup has a very favorable prognosis, with only a 10% probability of metastasis at diagnosis and a 5-year overall survival of 95–100%. SHH MBs carry a slightly greater risk of metastasis than WNT MBs but less than the Groups 3 and 4 subgroups. Prognosis of SHH tumors is inversely correlated with age, with 10-year overall survival of 77%, 51%, and 34% in infants, children, and adults, respectively.[
Non-WNT/SHH (Groups 3 and 4) MBs are more likely to be metastatic than WNT and SHH subgroups, with approximately 30% of the patients having metastasis at diagnosis. Group 4 tumors carry an intermediate prognosis with 5-year progression-free survival of 95%, with negative risk modifiers including MYCN amplification and presence of metastasis.[
A meta-analysis examining event-free survival/disease-free survival favored the inclusion of chemotherapy in treatment of pediatric MB when omitting, but not when including, disease progression as an event.[
Posterior fossa syndrome
The posterior fossa syndrome (PFS), also known as cerebellar mutism syndrome, is a complication that occurs in 8–24% of infratentorial brain tumor resections.[
While some studies have suggested vermian injury as the cause of PFS,[
CONCLUSION
MB is one of the most well-studied and frequently encountered CNS malignancies with a relatively good response to current treatments. However, there remains a subset of patients with poor outcomes despite numerous studies trying to optimize chemotherapy and radiation regimens. The current understanding of MB biology has vastly outpaced breakthroughs in treatment over the past 5–10 years, and application of this knowledge holds promise for continued improvements in outcomes for the future.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Aguilera D, Mazewski C, Fangusaro J, MacDonald TJ, McNall-Knapp RY, Hayes LL. Response to bevacizumab, irinotecan, and temozolomide in children with relapsed medulloblastoma: A multi-institutional experience. Childs Nerv Syst. 2013. 29: 589-96
2. Albright AL, Pollack IF, Adelson PD.editorsPrinciples and practice of pediatric neurosurgery. New York: Thieme; 2014. p.
3. Albright AL, Wisoff JH, Zeltzer PM, Boyett JM, Rorke LB, Stanley P. Effects of medulloblastoma resections on outcome in children: A report from the Children's Cancer Group. Neurosurgery. 1996. 38: 265-71
4. Avula S, Kumar R, Pizer B, Pettorini B, Abernethy L, Garlick D. Diffusion abnormalities on intraoperative magnetic resonance imaging as an early predictor for the risk of posterior fossa syndrome. Neuro Oncol. 2015. 17: 614-22
5. Avula S, Mallucci C, Kumar R, Pizer B. Posterior fossa syndrome following brain tumour resection: Review of pathophysiology and a new hypothesis on its pathogenesis. Childs Nerv Syst. 2015. 31: 1859-67
6. Baillieux H, Weyns F, Paquier P, De Deyn PP, Marien P. Posterior fossa syndrome after a vermian stroke: A new case and review of the literature. Pediatr Neurosurg. 2007. 43: 386-95
7. Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014. 40: 750-9
8. Brugieres L, Pierron G, Chompret A, Paillerets BB, Di Rocco F, Varlet P. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet. 2010. 47: 142-4
9. Coluccia D, Figuereido C, Isik S, Smith C, Rutka JT. Medulloblastoma: Tumor Biology and Relevance to Treatment and Prognosis Paradigm. Curr Neurol Neurosci Rep. 2016. 16: 43-
10. DeSouza RM, Jones BR, Lowis SP, Kurian KM. Pediatric medulloblastoma-Update on molecular classification driving targeted therapies. Front Oncol. 2014. 4: 176-
11. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005. 23: 7951-7
12. Gadgil N, Hansen D, Barry J, Chang R, Lam S. Posterior fossa syndrome in children following tumor resection: Knowledge update. Surg Neurol Int. 2016. 7: S179-83
13. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006. 7: 813-20
14. Giangaspero F, Perilongo G, Fondelli MP, Brisigotti M, Carollo C, Burnelli R. Medulloblastoma with extensive nodularity: A variant with favorable prognosis. J Neurosurg. 1999. 91: 971-7
15. Grill J, Sainte-Rose C, Jouvet A, Gentet JC, Lejars O, Frappaz D. Treatment of medulloblastoma with postoperative chemotherapy alone: An SFOP prospective trial in young children. Lancet Oncol. 2005. 6: 573-80
16. Grotzer MA, Janss AJ, Fung K, Biegel JA, Sutton LN, Rorke LB. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors. J Clin Oncol. 2000. 18: 1027-35
17. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM. The molecular basis of Turcot's syndrome. N Engl J Med. 1995. 332: 839-47
18. Kanamori M, Kumabe T, Saito R, Yamashita Y, Sonoda Y, Tominaga T. The safety of combination chemotherapy with ifosfamide, cisplatin, and etoposide (ICE): Single-institution retrospective review of 108 cases. No Shinkei Geka. 2010. 38: 997-1005
19. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012. 123: 473-84
20. Lam S, Reddy GD, Lin Y, Jea A. Management of hydrocephalus in children with posterior fossa tumors. Surg Neurol Int. 2015. 6: S346-8
21. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016. 530: 57-62
22. Lin J, Zheng Y, Chen K, Huang Z, Wu X, Zhang N. Inhibition of FOXM1 by thiostrepton sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. 2013. 30: 1739-44
23. McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007. 17: 151-64
24. Michiels EM, Schouten-Van Meeteren AY, Doz F, Janssens GO, van Dalen EC. Chemotherapy for children with medulloblastoma. Cochrane Database Syst Rev. 2015. 1: CD006678-
25. Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016. 529: 351-7
26. Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011. 122: 231-40
27. Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009. 41: 465-72
28. Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. 2009. 149A: 1539-43
29. Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009. 27: 1627-36
30. Puget S, Boddaert N, Viguier D, Kieffer V, Bulteau C, Garnett M. Injuries to inferior vermis and dentate nuclei predict poor neurological and neuropsychological outcome in children with malignant posterior fossa tumors. Cancer. 2009. 115: 1338-47
31. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016. 131: 821-31
32. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013. 14: 1200-7
33. Riva-Cambrin J, Detsky AS, Lamberti-Pasculli M, Sargent MA, Armstrong D, Moineddin R. Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr. 2009. 3: 378-85
34. Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005. 352: 978-86
35. Rutkowski S, von Bueren A, von Hoff K, Hartmann W, Shalaby T, Deinlein F. Prognostic relevance of clinical and biological risk factors in childhood medulloblastoma: Results of patients treated in the prospective multicenter trial HIT’91. Clin Cancer Res. 2007. 13: 2651-7
36. Saito R, Kumabe T, Sonoda Y, Kanamori M, Yamashita Y, Watanabe M. Combination chemotherapy with ifosfamide, cisplatin, and etoposide for medulloblastoma: Single-institute experience and differences in efficacy for subgroups of medulloblastoma. Childs Nerv Syst. 2011. 27: 1399-406
37. Slade I, Murray A, Hanks S, Kumar A, Walker L, Hargrave D. Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer. 2011. 10: 337-42
38. Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs). Cancer. 2012. 118: 1313-22
39. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002. 31: 306-10
40. Taylor MD, Mainprize TG, Rutka JT. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: A review. Neurosurgery. 2000. 47: 888-901
41. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012. 123: 465-72
42. Taylor RE, Bailey CC, Robinson K, Weston CL, Ellison D, Ironside J. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol. 2003. 21: 1581-91
43. Thompson EM, Hielscher T, Bouffet E, Remke M, Luu B, Gururangan S. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016. 17: 484-95
44. Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DM, Remke M. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015. 129: 449-57
45. Wells EM, Khademian ZP, Walsh KS, Vezina G, Sposto R, Keating RF. Postoperative cerebellar mutism syndrome following treatment of medulloblastoma: Neuroradiographic features and origin. J Neurosurg Pediatr. 2010. 5: 329-34
46. Wong TT, Liu YL, Ho DM, Chang KP, Liang ML, Chen HH. Factors affecting survival of medulloblastoma in children: The changing concept of management. Childs Nerv Syst. 2015. 31: 1687-98
47. Zhang ZY, Xu J, Ren Y, Li KK, Ng HK, Mao Y. Medulloblastoma in China: Clinicopathologic analyses of SHH, WNT, and non-SHH/WNT molecular subgroups reveal different therapeutic responses to adjuvant chemotherapy. PLoS One. 2014. 9: e99490-
48. Zurawel RH, Chiappa SA, Allen C, Raffel C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998. 58: 896-9

