

- Department of Neurosurgery, Shiga University of Medical Science, Otsu, Japan
- Department of Neurosurgery, Kusatsu General Hospital, Shiga, Japan
- Department of Neurosurgery, Koto Memorial Hospital, Shiga, Japan
Correspondence Address:
Naoki Nitta
Department of Neurosurgery, Shiga University of Medical Science, Otsu, Japan
DOI:10.4103/2152-7806.185781
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Nitta N, Nakasu S, Shima A, Nozaki K. mTORC1 signaling in primary central nervous system lymphoma. Surg Neurol Int 07-Jul-2016;7:
How to cite this URL: Nitta N, Nakasu S, Shima A, Nozaki K. mTORC1 signaling in primary central nervous system lymphoma. Surg Neurol Int 07-Jul-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/mtorc1-signaling-primary-central-nervous-system-lymphoma/
Abstract
Background:Mammalian target of rapamycin (mTOR) complex 1 (mTORC1) acts as a downstream effector of phosphatidyl-inositol-3 kinase, which is frequently hyperactivated in glioblastoma multiforme and links to cell signaling in cellular proliferation, differentiation, metabolism, and survival. Although many studies have suggested the importance of mTORC1 in tumorigenesis, its role remains unclear in brain tumors other than glioblastoma.
Methods:In the present study, we evaluated the activation of mTORC1 in 24 cases of primary central nervous system lymphoma (PCNSL).
Results:Immunohistochemical analysis showed overexpression of Rheb, which is immediately upstream of mTORC1, in 20 cases of PCNSL. Immunohistochemical analysis also showed overexpression of phospho-4E-BP1 (Thr37/46) and phospho-S6 (Ser235/236), which are increased after mTORC1 activation as mTORC1 downstream effectors in 17 and 21 cases, respectively.
Conclusion:Our data suggest that abnormal activation of the mTORC1 signaling pathway may cause tumor growth in patients with PCNSL.
Keywords: mTOR, phospho-4E-BP1, phospho-S6, primary central nervous system lymphoma, Rheb
INTRODUCTION
Mammalian target of rapamycin (mTOR) signaling impacts most major cellular functions, giving it an outsized role in regulating basic cell behaviors such as growth and proliferation.[
A small G protein Ras homologue enriched in brain (Rheb), regulated by a heterodimer consisting of tuberous sclerosis 1 (TSC1) and TSC2, directly interacts with mTORC1 and strongly stimulates its kinase activity when it is converted into its active GTP-bound state [
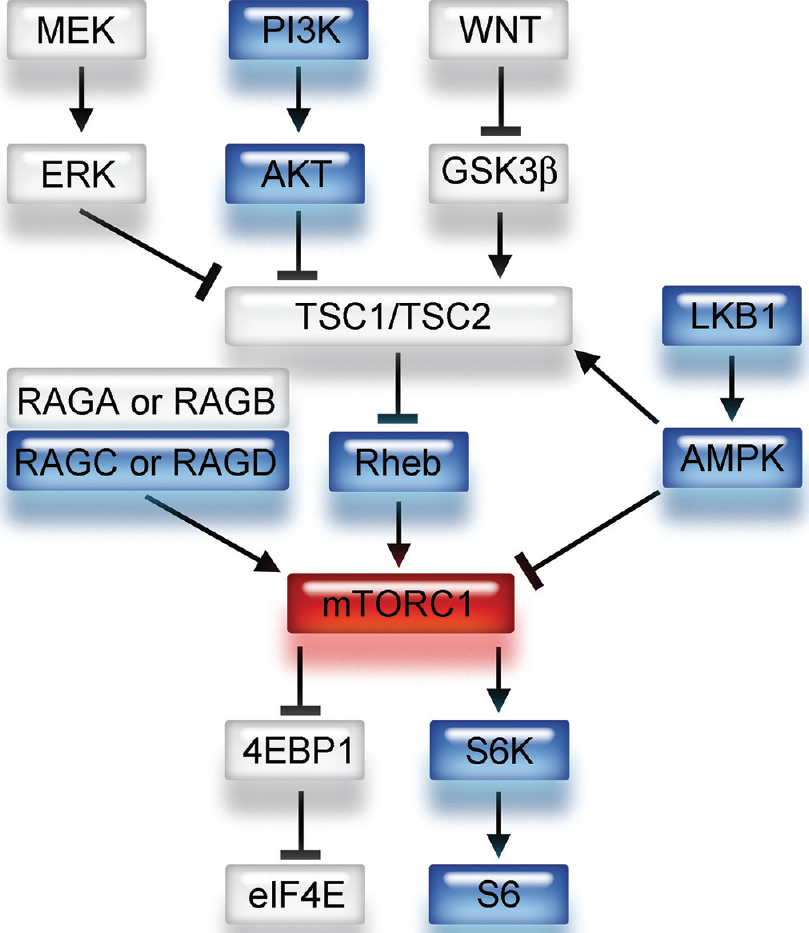
Figure 1
Schema of upstream and downstream signaling pathways of mTORC1. mTORC1 has several upstream pathways. Note that Rheb is a direct upstream component and 4E-BP1 and S6 are downstream components of the mTORC1 signaling pathway. GTP-bound Rheb activates mTORC1 and contributes to the phosphorylation of 4E-BP1 and S6
Primary central nervous system lymphomas (PCNSL) are estimated to account for approximately 3% of all CNS tumors. Despite high chemosensitivity and radiosensitivity and remissions being frequently of short duration, treatment of PCNSL remains challenging.[
MATERIALS AND METHODS
This retrospective study was approved by the Institutional Review Boards of Shiga University of Medical Science and was performed in accordance with the ethical standards laid down in the 1975 Declaration of Helsinki and its later amendments.
Tissue samples of consecutive cases of PCNSL (n = 27) obtained from January 1, 1998 to March 31, 2015 were collected from the surgical pathology files of the Shiga University of Medical Science Hospital, Shiga, Japan. All samples were obtained during the first surgery before chemotherapy and radiotherapy.
Sections of Formalin-fixed, paraffin-embedded tissues were stained with hematoxylin and eosin. Immunohistochemistry was performed for Rheb, which is an upstream regulator of mTORC1, and for p-4E-BP1 (Thr37/46) and p-S6 (Ser235/236), which are downstream effectors of mTORC1 [
Immunohistochemical staining quantification was categorized into two groups [low expressors, negative or focally positive (
RESULTS
In the normal brain tissues, Rheb was strongly expressed in neuronal soma in the cortex as “Ras homologue enriched in brain,” and weakly or not expressed in glial cells [
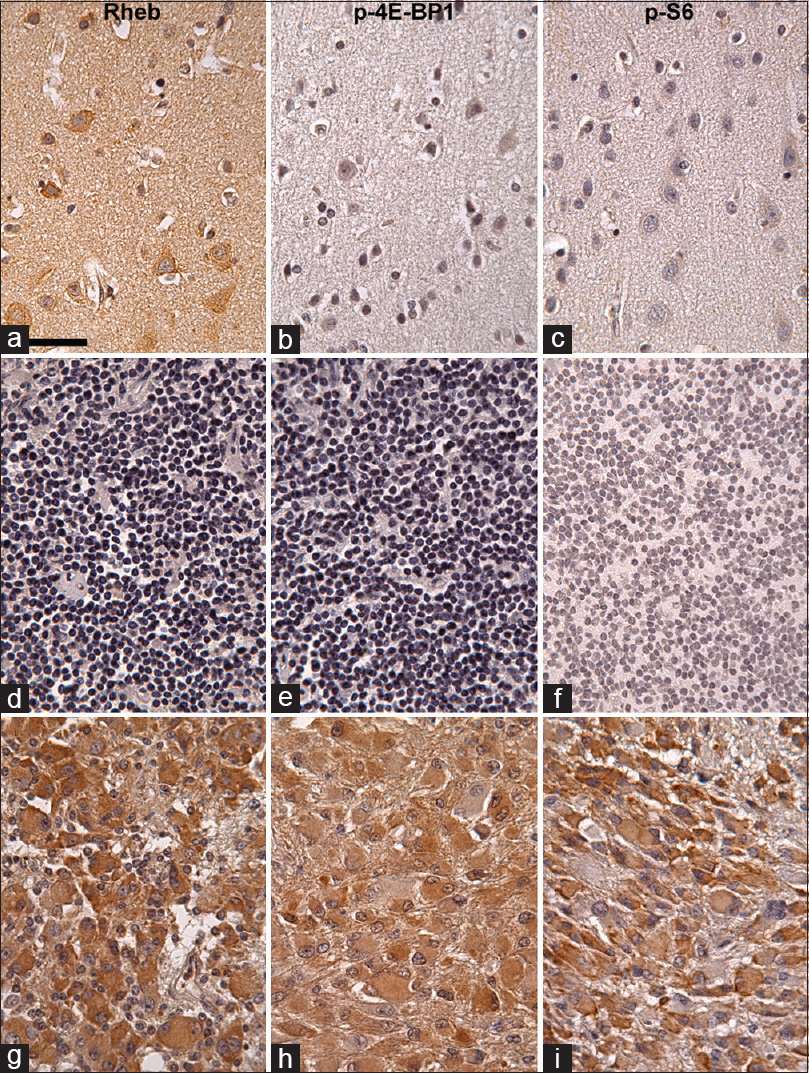
Figure 3
Rheb (a, d, g), p-4E-BP1 (b, e, h), and p-S6 (c, f, i) immunohistochemistry in the normal human cerebral cortex (a-c), normal lymph node (d-f), and SEGA (g-i) counterstained with hematoxylin. Most of the neuronal somas are strongly Rheb-positive in cytoplasm, not p-4E-BP1 positive, and mildly or not p-S6 positive (a-c). Glial cells are Rheb- p-4E-BP1-, and p-S6-negative (a-c). In normal lymph node, Rheb, p-4E-BP1, or p-S6 is not expressed (d-f). In SEGA, Rheb, p-4E-BP1, and p-S6 are moderately expressed in tumor cells, suggesting that the mTORC1 signaling pathway is activated in these cells. Scale bar = 50 μm in (a)

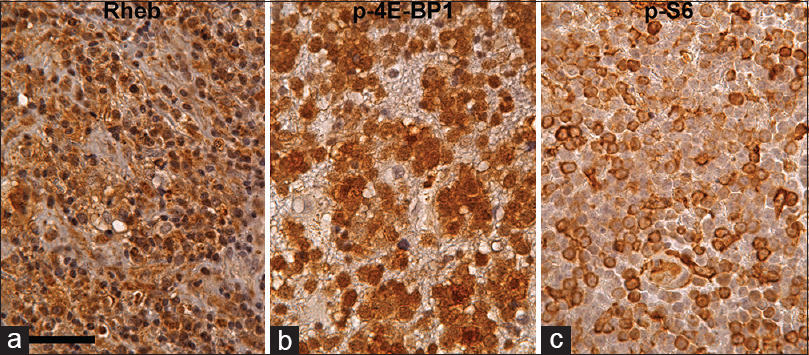
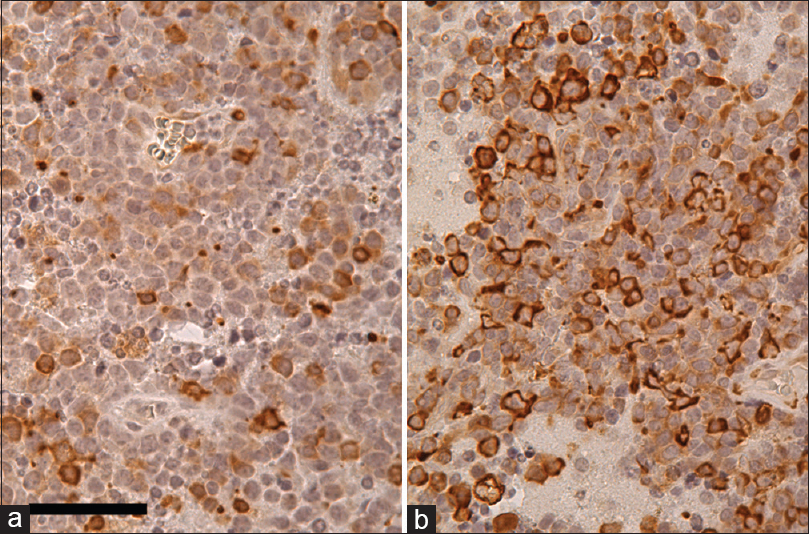
Figure 4
Rheb (a), p-4E-BP1 (b), and p-S6 (c) immunohistochemistry in PCNSL counterstained with hematoxylin. Most of the tumor cells are Rheb-positive in cytoplasm. Furthermore, some of the tumor cells are strongly p-4E-BP1 and p-S6 positive, suggesting that the mTORC1 signaling pathway is activated in these tumor cells. Scale bar = 50 μm in (a)
DISCUSSION
The present report describes activation of the mTORC1 signaling pathway in many cases of PCNSL. Both proliferation regulator p-4E-BP1 and cell size regulator p-S6 were highly expressed in half of cases of PCNSL, suggesting that mTORC1 activation contributes to not only rapid proliferation but also hypertrophy of the tumor cells of PCNSL.[
Lymphomas and mTORC1
Numerous studies have shown that the mTORC1 signaling pathway plays important roles in the biology of malignant cells, including deregulation of the cell cycle, suppression of apoptotic machinery and resistance to chemotherapies. The mTORC1 signaling pathway has been reported to be activated in systemic lymphomas, for example, Hodgkin lymphoma, anaplastic large cell lymphoma, diffuse large B-cell lymphoma, etc.[
Upstream markers of mTORC1 activation
In the present study, we evaluated mTORC1 activation using immunohistochemistry for Rheb, p-S6, and p-4E-BP1 because mTORC1 activity cannot be evaluated directly. Although many previous reports used expression of phospho-Akt (p-Akt) (Ser473) as an upstream marker of mTORC1 activation, recent studies have shown that there are several other upstream pathways of mTORC1, for example, mitogen-activated protein kinase signaling pathway, AMP-activated protein kinase signaling pathway, WNT signaling pathway, etc., [
Downstream markers of mTORC1 activation
mTORC1 directly phosphorylates translational regulators 4E-BP1 and S6K1, which, in turn, promote protein synthesis.[
mTORC1 activation in brain tumors
mTORC1 signaling pathway is activated in several brain tumors, in particular in gliomas, and increased expression of p-4E-BP1 and/or p-S6 in the tumors has been reported to correlate with enhancing proliferation and low survival.[
CONCLUSION
The mTORC1 signaling pathway is activated in PCNSL, which may contribute to proliferation and hypertrophy of tumor cells of PCNSL.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abel TW, Baker SJ, Fraser MM, Tihan T, Nelson JS, Yachnis AT. Lhermitte-Duclos disease: A report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTORpathway. J Neuropathol Exp Neurol. 2005. 64: 341-9
2. Annovazzi L, Mellai M, Caldera V, Valente G, Tessitore L, Schiffer D. mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 2009. 29: 3087-94
3. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012. 366: 520-9
4. Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: Biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004. 63: 1236-42
5. Cho D, Signoretti S, Dabora S, Regan M, Seeley A, Mariotti M. Potential histologic and molecular predictors of response to temsirolimus in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2007. 5: 379-85
6. Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A. Analysis of the phosphatidylinositol 3’-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003. 63: 2742-6
7. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006. 355: 1345-56
8. Drakos E, Rassidakis GZ, Medeiros LJ. Mammalian target of rapamycin (mTOR) pathway signalling in lymphomas. Expert Rev Mol Med. 2008. 10: e4-
9. Hoang-Xuan K, Bessell E, Bromberg J, Hottinger AF, Preusser M, Rudà R. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: Guidelines from the European Association for Neuro-Oncology. Lancet Oncol. 2015. 16: e322-32
10. Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010. 17: 249-61
11. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007. 356: 2271-81
12. Hütt-Cabezas M, Karajannis MA, Zagzag D, Shah S, Horkayne-Szakaly I, Rushing EJ. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro Oncol. 2013. 15: 1604-14
13. Im E, von Lintig FC, Chen J, Zhuang S, Qui W, Chowdhury S. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene. 2002. 21: 6356-65
14. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002. 4: 648-57
15. Iwenofu OH, Lackman RD, Staddon AP, Goodwin DG, Haupt HM, Brooks JS. Phospho-S6 ribosomal protein: A potential new predictive sarcoma marker for targeted mTOR therapy. Mod Pathol. 2008. 21: 231-7
16. James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO. NF2/merlin is a novel negative regulator of mTOR comple×1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009. 29: 4250-61
17. Jundt F, Raetzel N, Müller C, Calkhoven CF, Kley K, Mathas S. A rapamycin derivative (everolimus) controls proliferation through down-regulation of truncated CCAAT enhancer binding protein {beta} and NF-{kappa} B activity in Hodgkin and anaplastic large cell lymphomas. Blood. 2005. 106: 1801-7
18. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010. 363: 1801-11
19. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012. 149: 274-93
20. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997. 16: 64-7
21. Lu ZH, Shvartsman MB, Lee AY, Shao JM, Murray MM, Kladney RD. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 2010. 70: 3287-98
22. Martineau Y, Azar R, Bousquet C, Pyronnet S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013. 32: 671-7
23. Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008. 22: 2178-88
24. McBride SM, Perez DA, Polley MY, Vandenberg SR, Smith JS, Zheng S. Activation of PI3K/mTOR pathway occurs in most adult low-grade gliomas and predicts patient survival. J Neurooncol. 2010. 97: 33-40
25. McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011. 364: 1595-606
26. Montesinos-Rongen M, Siebert R, Deckert M. Primary lymphoma of the central nervous system: Just DLBCL or not?. Blood. 2009. 113: 7-10
27. Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008. 22: 2172-7
28. Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006. 66: 5618-23
29. Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005. 19: 2199-211
30. Shaw RJ, Cantley LC. Ras, PI (3) K and mTOR signalling controls tumour cell growth. Nature. 2006. 441: 424-30
31. Shimobayashi M, Hall MN. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014. 15: 155-62
32. Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003. 5: 559-65
33. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012. 485: 109-13
34. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009. 284: 8023-32
35. Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001. 276: 38052-60
36. Wagner AJ, Malinowska-Kolodziej I, Morgan JA, Qin W, Fletcher CD, Vena N. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: Targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010. 28: 835-40
37. Wolff N, Kabbani W, Bradley T, Raj G, Watumull L, Brugarolas J. Sirolimus and temsirolimus for epithelioid angiomyolipoma. J Clin Oncol. 2010. 28: e65-8
38. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011. 364: 514-23
39. Zoncu R, Efeyan A, Sabatini DM. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011. 12: 21-35

