

- Department of Neurological Surgery, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA
- Department of Pathology, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA
- Department of Radiology, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA
Correspondence Address:
Ian F. Dunn
Department of Neurological Surgery, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA
DOI:10.4103/sni.sni_7_17
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: David L. Penn, Richard J. Tartarini, Carolyn H. Glass, Umberto De Girolami, Amir A. Zamani, Ian F. Dunn. Natural history of cranial fibrous dysplasia revealed during long-term follow-up: Case report and literature review. 06-Sep-2017;8:209
How to cite this URL: David L. Penn, Richard J. Tartarini, Carolyn H. Glass, Umberto De Girolami, Amir A. Zamani, Ian F. Dunn. Natural history of cranial fibrous dysplasia revealed during long-term follow-up: Case report and literature review. 06-Sep-2017;8:209. Available from: http://surgicalneurologyint.com/surgicalint-articles/natural-history-of-cranial-fibrous-dysplasia-revealed-during-long%e2%80%91term-follow%e2%80%91up-case-report-and-literature-review/
Date of Submission
05-Jan-2017
Date of Acceptance
04-May-2017
Date of Web Publication
06-Sep-2017
Abstract
Background:Fibrous dysplasia (FD) is a rare developmental disease characterized by the replacement of bone marrow with proliferating fibro-osseous tissue. There exist three forms of FD—monostotic, polyostotic, and that associated with McCune-Albright syndrome. The disease can present in different locations and with a variety of symptoms. One of the more common locations of FD occurrence is the craniofacial region. Treatment of asymptomatic FD often involves conservative management with serial imaging. Medical management with bisphosphonates is an option, though long-term efficacy data are lacking. Surgical resection is usually reserved for very large or symptomatic lesions.
Case Description:We discuss the most unusual case of a 52-year-old male found to have a left pterional mass while being worked up for sinus headaches. The patient elected to follow this lesion conservatively, and imaging several years later showed obvious growth which accelerated in the last 4 years during an 18-year observational period. He ultimately underwent successful resection of an extradural and intradural FD.
Conclusions:The significant growth potential of these lesions was revealed in this patient, in whom conservative management had been adopted. Despite optimal surgical resection and outcome in this case, the importance of surveillance imaging and perhaps earlier intervention cannot be underestimated when managing cranial FD.
Keywords: Craniofacial, fibrous dysplasia, middle fossa zygomatic approach, polyostotic
INTRODUCTION
Fibrous dysplasia (FD) is a rare developmental disease characterized by the replacement of normal bone marrow with proliferating fibro-osseous tissue and thinning of cortical bone that may cause deformities, pain, swelling, loss of coordination, and/or nerve compression.[
FD, if asymptomatic, is often managed conservatively. Medical therapy with bisphosphonates is controversial, having been previously shown to result in significant decreases in pain and radiographic lesions; however, a recent randomized, double blind, placebo-controlled trial disputes these effects.[
Here, we present a unique case of presumed radiographic FD followed conservatively for 18 years. Its growth rate, symptom onset, and findings of intradural invasion confer important insights into the natural history of FD and may suggest an earlier role for surgery in select cases.
CASE DESCRIPTION
The patient is a 52-year-old right-handed gentleman, who initially presented in 1998 with headaches that were thought to be related to sinus infections. Initial work up consisted of X-ray of the skull, which demonstrated a dense calcified lesion in the left frontotemporal region [
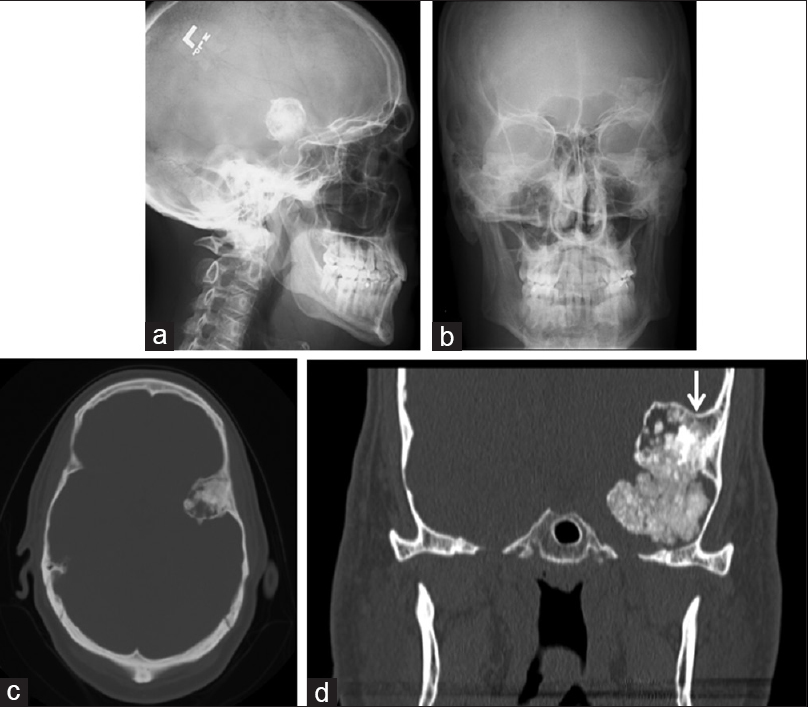
Figure 1
(a and b) (Lateral and AP views of skull) and (c and d) (axial and coronal CT) show a heavily calcified lesion in the left frontotemporal region with its origin from the temporal bone immediately behind the pterion. The mass is non-homogeneous with sharply defined margins. The arrow indicates where the lesion may have become intradural
The patient presented again in 2014 with similar headaches, again thought to be caused by sinus infection. At this time, an otolaryngologist ordered a computed tomography (CT) scan, which demonstrated growth of the mass [
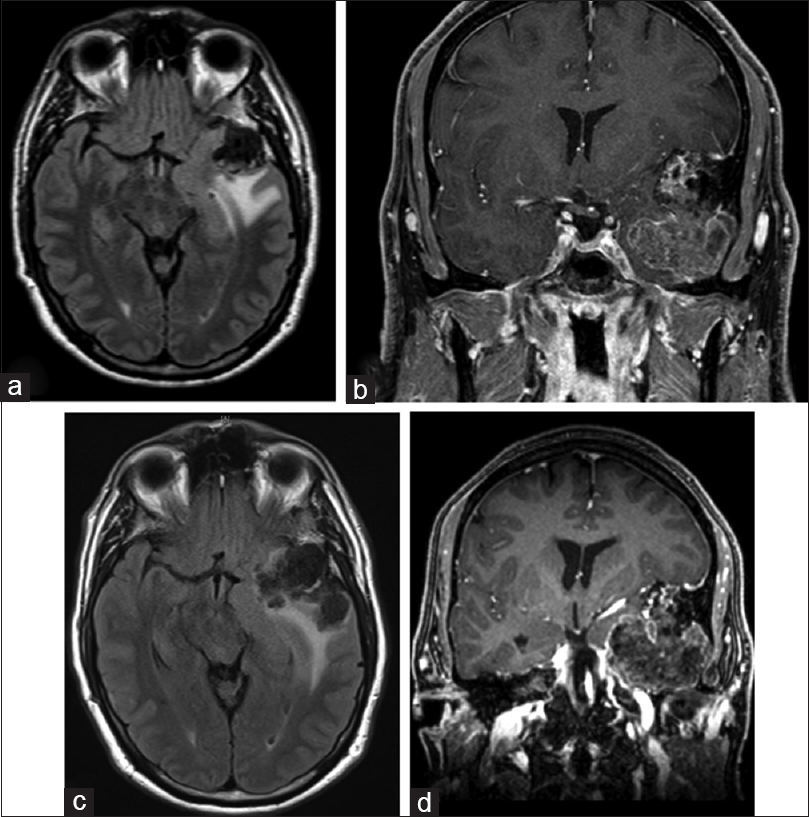
Figure 2
MR images (a and b) are from 2014; (c and d) are from 2016, just prior to surgical resection. These demonstrate growth of this lesion over a two-year period. Axial FLAIR images (a and c) demonstrate adjacent vasogenic edema extending in the left temporal lobe and optic radiation. These images reveal the extensive nature of this mass, occupying a large part of the left middle cranial fossa with resultant shift of the uncus medially deforming the suprasellar cistern and shifting the midbrain. On coronal images upward displacement of left MCA is seen
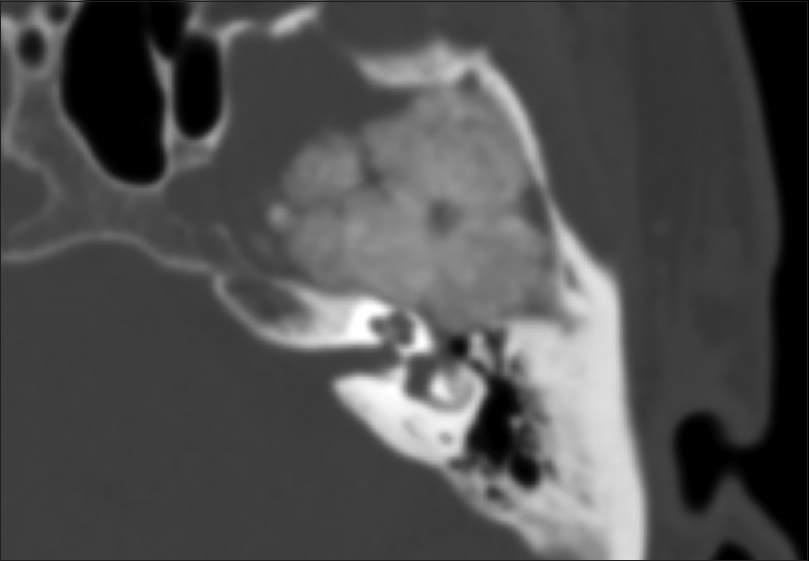
CT angiography (CTA) was obtained demonstrating close proximity of the mass to the left cavernous and ophthalmic internal carotid artery, as well as M2 branches of the middle cerebral artery (MAC), without obvious stenosis, involvement, or entrapment of these vessels. While his hearing was intact and face symmetric, there was an unclear relationship of the mass to the geniculate ganglion and cochlea on immediate preoperative imaging studies [
In 2016, approximately 18 years after the discovery of this mass, the patient underwent surgery for resection of this lesion through a frontotemporal craniotomy and craniectomy, and zygomatic osteotomy to maximize our access to the middle fossa floor. We sectioned the zygoma and reflected the temporalis inferiorly after subfascial dissection to gain adequate access to the floor of the middle fossa. We used neuronavigation to identify the mass emanating from the inner table, placing burr holes immediately around this region and connecting these. This allowed us to separate the mass from the larger frontotemporal craniotomy flap, which we then elevated. We used a high-speed drill to remove the extradural portion of the mass which at the superficial depth of the sylvian fissure became intradural. The MCA branches were easily dissected from the lesion and the middle fossa dura found to be intact. After a complete resection we replaced the frontotemporal flap, reconstructing the central portion with titanium mesh. Postoperative imaging showed a complete resection [
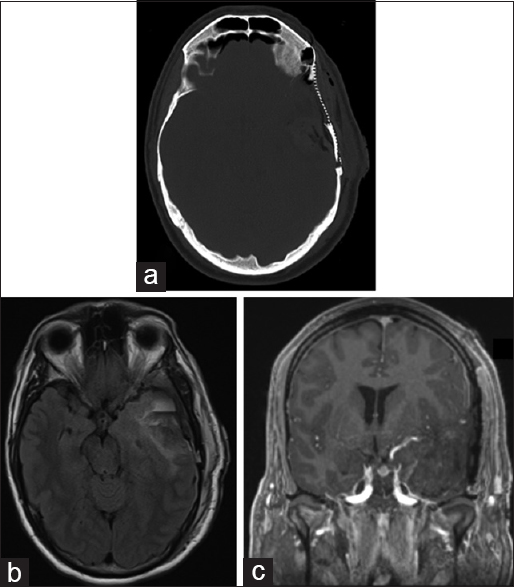
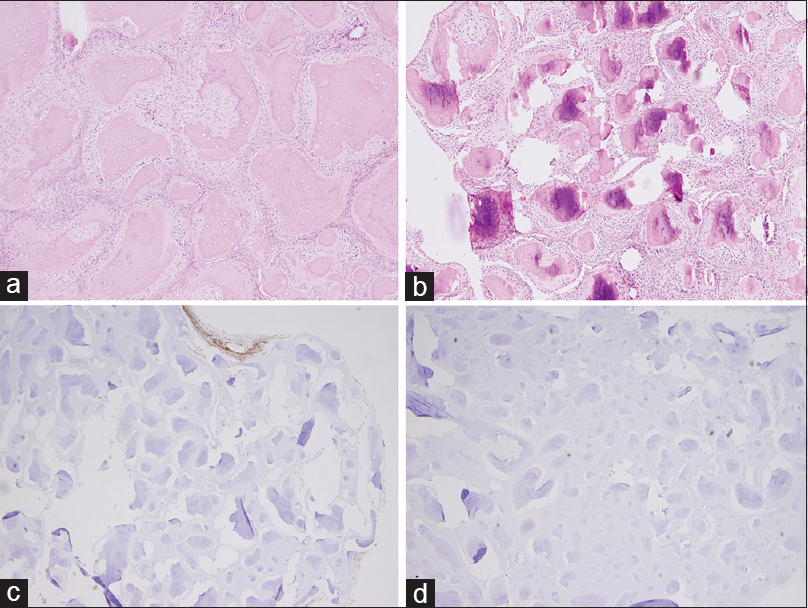
Figure 5
Permanent pathology obtained after surgical resection of the lesion. Panel (a and b) represent H&E stained sections demonstrating curvilinear trabeculae of woven bone surrounded by moderately cellular fibrous stroma. Negative epithelial membrane antigen stain and negative somatostatin receptor 2 staining are represented in Panel (c and d), respectively
DISCUSSION
The natural history of FD, as well as the manifestation of symptoms, is not well understood, making it difficult for clinicians to develop standardized treatment plans.[
In general, surgical treatment is not necessary in asymptomatic cases of FD; however, surgery is a consideration when lesions become symptomatic, grow, or exert mass effect. Many authors view radical surgical resection as the most successful and definitive treatment over more conservative procedures.[
Amit et al. performed a case series of 241 patients with cranial FD and optic nerve involvement (386 optic nerves investigated) to assess the outcomes of surgical treatment versus conservative management.[
Another critical consideration is the timing of surgical intervention. Growth of lesions can serve as an indicator for future symptoms. In addition to our patient, several cases reported in the literature have involved monthly growth with worsening symptoms appearing closer to surgical intervention.[
Radiographically, FD typically has an asymmetric ground-glass appearance on CT scan. FD blends with normal bone and results in thinning of the cortical bone.[
CONCLUSIONS
FD is a developmental disease characterized by the replacement of normal bone marrow with proliferating fibro-osseous tissue, presenting in multiple forms and with a variety of symptoms. One of the most common places for FD to occur is the craniofacial region, in particular the frontal bone, presenting with symptoms including headaches or cranial nerve deficits. The present study follows an initially asymptomatic patient with a growing, atypical left frontotemporal FD lesion clinically and radiographically over 18 years, providing some insight into the natural history of cranial FD. Although treatment strategies are controversial, complete surgical resection of large cranial FD lesions remains a viable plan for successful management of this disease. In this case, deferring surgical resection did not adversely affect surgical resection and outcome, but many characteristics of the lesion should be accounted for when creating a treatment plan.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Adada B, Al-Mefty O. Fibrous dysplasia of the clivus. Neurosurgery. 2003. 52: 318-22
2. Adetayo OA, Salcedo SE, Borad V, Richards SS, Workman AD, Ray AO. Fibrous dysplasia: An overview of disease process, indications for surgical management, and a case report. Eplasty. 2015. 15: e6-
3. Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM, Gil Z. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia-a meta-analysis. PLoS One. 2011. 6: e25179-
4. Amit M, Fliss DM, Gil Z. Fibrous dysplasia of the sphenoid and skull base. Otolaryngol Clin North Am. 2011. 44: 891-
5. Benhamou J, Gensburger D, Messiaen C, Chapurlat R. Prognostic Factors from an Epidemiologic Evaluation of Fibrous Dysplasia of Bone in a Modern Cohort. The Francedys Study. J Bone Miner Res. 2016. 31: 2167-72
6. Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014. 99: 4133-40
7. Chapurlat RD, Hugueny P, Delmas PD, Meunier PJ. Treatment of fibrous dysplasia of bone with intravenous pamidronate: Long-term effectiveness and evaluation of predictors of response to treatment. Bone. 2004. 35: 235-42
8. Chen YR, Noordhoff MS. Treatment of craniomaxillofacial fibrous dysplasia: How early and how extensive?. Plast Reconstr Surg. 1990. 86: 835-
9. Cohen MM, Howell RE. Etiology of fibrous dysplasia and McCune-Albright syndrome. Int J Oral Maxillofac Surg. 1999. 28: 366-71
10. Cutler CM, Lee JS, Butman JA, FitzGibbon EJ, Kelly MH, Brillante BA. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: Risk factors for blindness and safety of observation. Neurosurgery. 2006. 59: 1011-
11. DiCaprio MR, Enneking WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005. 87: 1848-64
12. Gupta A, Mehta VS, Sarkar C. Large cystic fibrous dysplasia of the temporal bone: Case report and review of literature. J Clin Neurosci. 2003. 10: 364-7
13. Happle R. The McCune-Albright syndrome: A lethal gene surviving by mosaicism. Clin Genet. 1986. 29: 321-4
14. Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012. 7: S2-
15. Munro IR, Chen YR. Radical treatment for fronto-orbital fibrous dysplasia: The chain-link fence. Plast Reconstr Surg. 1981. 67: 719-30
16. Muthusamy S, Subhawong T, Conway SA, Temple HT. Locally aggressive fibrous dysplasia mimicking malignancy: A report of four cases and review of the literature. Clin Orthop Relat Res. 2015. 473: 742-50
17. Ozek C, Gundogan H, Bilkay U, Tokat C, Gurler T, Songur E. Craniomaxillofacial fibrous dysplasia. J Craniofac Surg. 2002. 13: 382-9
18. Pardo-Maza A, Lassaletta L, Ruiz-Bravo E, Perez-Mora R, Penarrocha J, Gavilan J. Fibrous dysplasia of the temporal bone secondary to ear surgery: A case report. J Med Case Rep. 2015. 9: 129-
19. Park B, Abode-Iyamah K, Lee SL, Kirby P, El-Khoury G, Wilson S. Fibro-osseous lesion of the cranium in an adolescent patient. Surg Neurol Int. 2015. 6: 12-
20. Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003. 88: 4569-75
21. Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: A case series and literature review. J Oral Maxillofac Surg. 2001. 59: 157-
22. Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994. 73: 1411-24
23. Sadeghi SM, Hosseini SN. Spontaneous conversion of fibrous dysplasia into osteosarcoma. J Craniofac Surg. 2011. 22: 959-61
24. Tan YC, Yu CC, Chang CN, Ma L, Chen YR. Optic nerve compression in craniofacial fibrous dysplasia: The role and indications for decompression. Plast Reconstr Surg. 2007. 120: 1957-62
25. Turner OA. Fibrous dysplasia of bone; neurosurgical considerations in two cases. Yale J Biol Med. 1951. 23: 501-4
26. Unal Erzurumlu Z, Celenk P, Bulut E, Baris YS. CT Imaging of Craniofacial Fibrous Dysplasia. Case Rep Dent 2015. 2015. p. 134123-
27. Valentini V, Cassoni A, Marianetti TM, Terenzi V, Fadda MT, Iannetti G. Craniomaxillofacial fibrous dysplasia: Conservative treatment or radical surgery. A retrospective study on 68 patients?. Plast Reconstr Surg. 2009. 123: 653-60
28. Weinstein LS, Chen M, Liu J. Gs (alpha) mutations and imprinting defects in human disease. Ann N Y Acad Sci. 2002. 968: 173-97
29. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991. 325: 1688-95
30. Yuceer N, Kutluhan A, Bekerecioglu M, Arslan H, Akman E. Polyostotic fibrous dysplasia with craniofacial localization presenting with frontal lobe compression in a 14-year-old girl. Acta Neurochir (Wien). 1999. 141: 203-7

