

- Department of Clinical Neuropathology, King's College Hospital NHS Foundation Trust, London, UK
- Department of Neurosurgery, King's College Hospital NHS Foundation Trust, London, UK
- Department of Neuroradiology, King's College Hospital NHS Foundation Trust, London, UK
Correspondence Address:
Istvan Bodi
Department of Neurosurgery, King's College Hospital NHS Foundation Trust, London, UK
DOI:10.4103/sni.sni_334_17
Copyright: © 2018 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Istvan Bodi, Anastasios Giamouriadis, Naomi Sibtain, Ross Laxton, Andrew King, Francesco Vergani. Primary intracerebral INI1-deficient rhabdoid tumor with CD34 immunopositivity in a young adult. 21-Feb-2018;9:45
How to cite this URL: Istvan Bodi, Anastasios Giamouriadis, Naomi Sibtain, Ross Laxton, Andrew King, Francesco Vergani. Primary intracerebral INI1-deficient rhabdoid tumor with CD34 immunopositivity in a young adult. 21-Feb-2018;9:45. Available from: http://surgicalneurologyint.com/surgicalint-articles/primary-intracerebral-ini1%e2%80%91deficient-rhabdoid-tumor-with-cd34-immunopositivity-in-a-young-adult/
Date of Submission
08-Sep-2017
Date of Acceptance
12-Oct-2017
Date of Web Publication
21-Feb-2018
Abstract
Background:Primary CNS malignant rhabdoid tumors are very rare in adults and much less is known about their biological behavior than in children. Recently, two adult cases of SMARCB1 (also known as INI1)-deficient tumor with rhabdoid cells have been described, suggesting an emerging group of primary meningeal SMARCB1-deficient tumors. We have recently encountered a case of INI1-deficient tumor with similar histology and immunophenotype to the above cases, but with a superficial cerebral, yet apparent intra-axial origin.
Case Description:A 22-year-old woman presented with approximately one year history of focal sensorimotor right upper limb seizures and recently developed a slowly progressive weakness in her right hand. An MRI of the brain demonstrated an avidly enhancing lesion centered on the left perirolandic region with no definite dural involvement. The patient underwent a complete surgical excision. Histology revealed a tumor with monotonous epithelioid and spindle-shaped cells in a mucoid/myxoid background. There was focal mitotic activity and a few necrotic areas, in addition to many rhabdoid cells. The immunohistochemistry was negative for INI1 and there was strong positivity with CD34, while focal smooth muscle actin (SMA) and epithelial membrane antigen (EMA) immunoreactivity were also noted.
Conclusions:As an addition to the two cases of adult SMARCB1-deficient tumors recently described, we present a further adult case with a similar immunohistochemical profile but with an apparent intra-axial origin, questioning the necessary meningeal origin of this type of tumor. The prognosis of this adult INI1/SMARCB1-deficient tumor is to be determined, but may be better than the pediatric atypical/teratoid tumor (AT/RT).
Keywords: Atypical teratoid/rhabdoid tumor, CD34, INI1, malignant rhabdoid tumor, SMARCB1
INTRODUCTION
Atypical teratoid/rhabdoid tumour (AT/RT) is a malignant CNS embryonal tumour composed of poorly differentiated cells with immunohistochemically polyphenotypical differentiation along neuroectodermal, epithelial, and mesenchymal lines frequently containing rhabdoid cells.[
Recently, Dadone et al. described two adult cases of SMARCB1 (also known as INI1, hSNF5, and BAF47)-deficient tumor.[
CASE DESCRIPTION
A 22-year-old woman with no significant medical background presented with approximately one year history of focal sensorimotor right upper limb seizures, characterised by occasional twitching and pins and needles in the right hand and forearm. Over the previous two months she also developed a slowly progressive weakness in her right hand. Seizure activity was well controlled on monotherapy with levetiracetam. An MRI of the brain demonstrated an avidly enhancing lesion centered in the left perirolandic region (involving precentral and postcentral gyri) measuring 38 × 43 × 39 mm [Figure
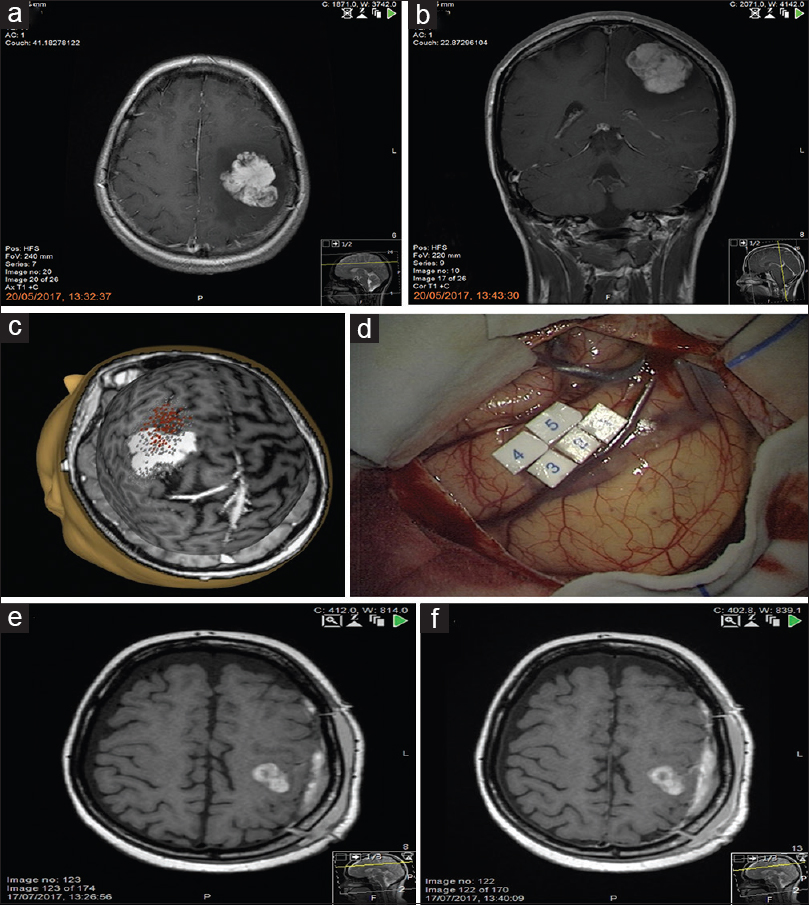
Figure 1
Pre-operative MRI (a, b) T1-weighted post gadolinium images show an avidly enhancing mass with well-defined, lobulated borders in the left frontoparietal region. Preoperative transcranial magnetic stimulation (c) and intraoperative motor mapping and monitoring (d) demonstrate that the primary motor cortex was located in front of the tumor, confirming the location of the tumor within the central sulcus. Post-operative MRI axial T1-weighted pre gadolinium (e) and post gadolinium (f) show a small volume of hemorrhage in the surgical bed but no residual tumor
The toluidine blue stained intraoperative smear preparation confirmed neoplastic tissue with a loose matrix and monomorphic tumor cells together with associated mast cells but no definite diagnosis could be given [
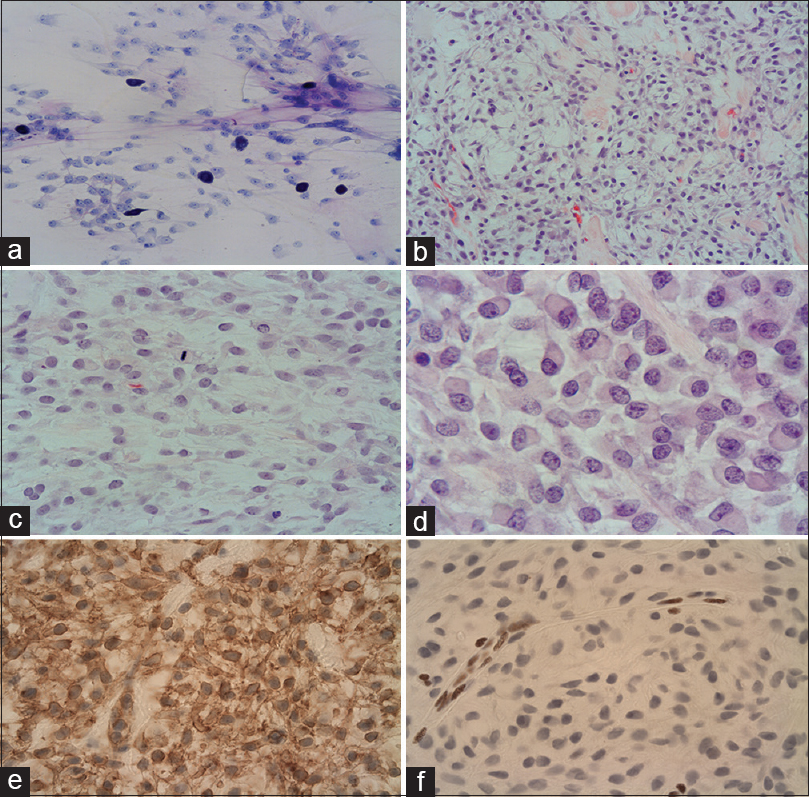
Figure 2
Toluidine blue stained smear preparation (a) shows spindle shaped, monomorphic neoplastic cells with prominent nucleoli in a loose matrix and mast cells. Epithelioid and spindle-shaped cells arranged in mucoid/myxoid background with hyalinised elements (b, c). The cytoplasm is frequently vacuolated and a mitotic figure is noted (c). Eosinophilic cytoplasmic inclusions in keeping with rhabdoid cells (d). Strong, diffuse immunoreactivity with CD34 (e). Loss of expression in the tumor cells by INI1, while the nuclear staining is preserved in the endothelial cells (f)
Immunohistochemistry showed strong, diffuse positivity with vimentin, synaptophysin, CD34 [
There was no evidence of mutations of BRAF V600E, IDH1, and IDH2 by pyrosequencing methods from genomic DNA extracted from formalin fixed paraffin embedded tissue. There was also no evidence of a KIAA1549-BRAF gene fusion by RT-PCR analysis. The MGMT promoter was unmethylated.
DISCUSSION
The presence of rhabdoid cells and the lost INI1 (BAF47) expression in an intracranial location would usually confirm the diagnosis of atypical teratoid/rhabdoid tumor (AT/RT).[
We have reviewed our departmental database going back to 1995 for AT/RT and performed immunohistochemistry in selected cases if necessary to confirm the diagnosis. Altogether we found 10 cases of INI1-deficient tumors [unpublished results]; the majority occurred in early childhood (age range 4 months to 2 years) and only two occurred in adults (49 year old female patient with pituitary AT/RT and the current case).[
The prognosis of the adult intracranial AT/RT is still to be determined. A recent literature review of 50 adult AT/RT cases found the median time to progression was 5 months and the overall survival was 23 months.[
Our current case also occurred in a peripheral location of the frontal lobe with no connection to the ventricular system and the surgical resection appeared to be complete. Moreover the mitotic activity was low and the proliferation activity was moderate, corresponding to the relatively long history of 15 months prior surgery in our case. Post-operatively the patient recovered with normal power in her hand. Recurrent focal seizure activity was controlled increasing the dose of levetiracetam. Whole neuraxis MRI was performed after the histology result for disease staging and no spinal secondary deposit was identified [Figure
CONCLUSIONS
In conclusion, as an addition to the two cases of adult SMARCB1-deficient tumors recently described, we further present an adult case with a similar immunohistochemical profile but with an apparent intra-axial origin, questioning the necessary meningeal origin of this type of tumor. The prognosis of this adult INI1/SMARCB1-deficient tumor is to be determined, but may be better than the pediatric AT/RT.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that name and initial will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
Informed consent was obtained from the patient for publication.
References
1. Almalki MH, Alrogi A, Al-Rabie A, Al-Dandan S, Altwairgi A, Orz Y. Atypical Teratoid/Rhabdoid Tumor of the Sellar Region in an Adult With Long Survival: Case Report and Review of the Literature. J Clin Med Res. 2017. 9: 216-20
2. Bello L, Riva M, Fava E, Ferpozzi V, Castellano A, Raneri F. Tailoring neurophysiological strategies with clinical context enhances resection and safety and expands indications in gliomas involving motor pathways. Neuro Oncol. 2014. 16: 1110-28
3. Bodi I, Poitelea M, Bullock P, Brazil L, Hing SN, Jacques TS, King A. 113th Meeting of the British Neuropathological Society. Neuropathol Appl Neurobiol. 2011. 37: 810-1
4. Cha YJ, Hong C-K, Kim D-S, Lee S-K, Park HJ, Kim SH. Poorly differentiated chordoma with loss of SMARCB1/INI1 expression in pediatric patients: A report of two cases and review of the literature. Neuropathology. 2017. p.
5. Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. 2007. 84: 217-22
6. Dadone B, Fontaine D, Mondot L, Cristofari G, Jouvet A, Godfraind C. Meningeal SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily B member 1 (SMARCB1) -deficient tumours: An emerging group of meningeal tumours. Neuropathol Appl Neurobiol. 2017. 1: 433-49
7. Dardis C, Yeo J, Milton K, Ashby LS, Smith KA, Mehta S. Atypical teratoid rhabdoid tumor: Two case reports and an analysis of adult cases with implications for pathophysiology and treatment. Front Neurol. 2017. 8: 1-14
8. Frühwald MC, Biegel JA, Bourdeaut F, Roberts CWM, Chi SN. Atypical teratoid/rhabdoid tumors-Current concepts, advances in biology, and potential future therapies. Neuro Oncol. 2016. 18: 764-78
9. Hollmann TJ, Hornick JL. INI1-Deficient Tumors. Am J Surg Pathol. 2011. 35: e47-e63
10. Horiguchi H, Nakata S, Nobusawa S, Uyama S, Miyamoto T, Ueta H. Adult-onset atypical teratoid/rhabdoid tumor featuring long spindle cells with nuclear palisading and perivascular pseudorosettes. Neuropathology. 2017. 37: 52-7
11. Ibrahim GM, Huang A, Halliday W, Dirks PB, Malkin D, Baskin B. Cribriform neuroepithelial tumour: Novel clinicopathological, ultrastructural and cytogenetic findings. Acta Neuropathol. 2011. 122: 511-4
12. Louis DN, Ohgaki H, Wiestler OD. WHO classification of tumours of the central nervous system. International Agency For Research On Cancer. 2016. 131: 803-20
13. Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: Review of four cases. J Neurooncol. 2008. 88: 321-30
14. Nobusawa S, Hirato J, Sugai T, Okura N, Yamazaki T, Yamada S. Atypical Teratoid/Rhabdoid Tumor (AT/RT) arising from ependymoma: A type of AT/RT secondarily developing from other primary central nervous system tumors. J Neuropathol Exp Neurol. 2016. 75: 167-74
15. Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. 2005. 15: 23-8
16. Umredkar A, Bal A, Vashista RK. Atypical teratoid/rhabdoid tumour of the central nervous system in adult: Case report. Br J Neurosurg. 2010. 24: 699-704
17. Uner M, Saglam A, Meydan BC, Aslan K, Soylemezoglu F. Atypical teratoid rhabdoid tumor arising in a pleomorphic xanthoastrocytoma: A rare entity. Clin Neuropathol. 2017. 36: 227-32

