

- Department of Neurosurgery, Institute for Neurological Research FLENI, Buenos Aires, Argentina
- Department of Pathology, Institute for Neurological Research FLENI, Buenos Aires, Argentina
Correspondence Address:
J. F. Villalonga
Department of Neurosurgery, Institute for Neurological Research FLENI, Buenos Aires, Argentina
DOI:10.4103/sni.sni_490_16
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: A. Cervio, J. F. Villalonga, R. Mormandi, S. Condomí Alcorta, G. Sevlever, J. Salvat. Surgical treatment of cerebellar hemangioblastomas. 01-Aug-2017;8:163
How to cite this URL: A. Cervio, J. F. Villalonga, R. Mormandi, S. Condomí Alcorta, G. Sevlever, J. Salvat. Surgical treatment of cerebellar hemangioblastomas. 01-Aug-2017;8:163. Available from: http://surgicalneurologyint.com/surgicalint-articles/surgical-treatment-of-cerebellar-hemangioblastomas/
Date of Submission
21-Dec-2016
Date of Acceptance
21-May-2017
Date of Web Publication
01-Aug-2017
Abstract
Background:Hemangioblastomas (HBL) are uncommon tumors of the central nervous system (CNS), corresponding to 1–2.5% of all intracranial tumors. They can present sporadically or in patients with von Hippel–Lindau (VHL) disease and are most often located in the cerebellum, brainstem, and spinal cord. VHL disease is a multiple neoplasia syndrome inherited in an autosomal dominant fashion and caused by a VHL suppressor gene deletion. We present our experience in the management of patients with cerebellar HBL.
Methods:Thirty consecutive patients with cerebellar HBL were included in this study. Hospital charts, radiological images, and operative records were reviewed. Modified Rankin scores were used to evaluate the clinical course.
Results:Thirty patients diagnosed with cerebellar HBL were operated. Complete total resection was achieved in 93% of the cases. Postoperatively, 83% of the patients showed good functional recovery.
Conclusions:HBL of the cerebellum should be resected when symptomatic or when the tumor (or a tumor-associated cyst) shows signs of enlargement. Surgical intent should seek en bloc resection to minimize intraoperative bleeding. Patients with HBLs must be tested for VHL gene mutations, and in confirmed cases, relatives should be offered genetic counseling.
Keywords: Cerebellum, hemangioblastomas, microsurgery, von Hippel–Lindau disease
INTRODUCTION
Hemangioblastomas (HBL) are uncommon tumors of the central nervous system (CNS) corresponding to 1–2.5% of all intracranial tumors.[
We present our experience with the treatment of sporadic cerebellar HBLs as well as those associated with VHL disease, analyzing patients’ clinical presentation, imaging characteristics, surgical findings, postoperative complications, and long-term follow-up.
MATERIALS AND METHODS
A retrospective review was conducted using medical charts of patients subjected to surgical treatment for cerebellar HBL at our institution between January 1995 and December 2014.
The information obtained included patient demographics, personal history, clinical presentation, postoperative course, long-term follow-up, as well as imaging studies [magnetic resonance imaging (MRI) with T1, T2, fluid attenuated inversion recovery (FLAIR), and gradient echo (GRE) sequences, with and without contrast]. Preoperative images confirmed whether lesions were single or multiple, cystic or solid, and whether edema and/or bleeding were present. Surgical notes provided details regarding the use of preoperative embolization, type of surgical approach, operative procedure, as well as presence and degree of tumor bleeding. Postoperative complications and long-term clinical course were established during patient follow-up. MRI, starting 3 months after surgery, showed extension of tumor resection (total or subtotal), new lesion development, and clinical course in cases of unresected cysts. The modified Rankin scale (mRS) was used to rate functional independence, first in the immediate postoperative period, and then on a yearly basis [

VHL disease diagnosis was established based on the updated criteria described by Conway et al.[

Statistical analysis
Data was analyzed using STATA: Version 12 (StataCorp. 2011. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP).
The studies were approved by the ethics committee of our institution, and informed consent was obtained from each patient.
RESULTS
Thirty case histories of patients presenting with cerebellar HBL were identified. Sixteen corresponded to males (53%) and 14 to females (47%). In 26 cases (87%), the lesions were sporadic, whereas VHL diagnosis was confirmed in 4 patients (13%). Conway criteria were applied for VHL diagnosis in 3 patients, and genetic sequencing from a peripheral blood sample confirmed the presence of gene deletion in 1 patient. Twenty-eight cases presented single lesions (93%) and 2 cases multiple lesions (7%). Sporadic HBLs (n = 26) included 13 (50%) cystic lesions and 13 solid tumors (50%). Thirty-seven percent of patients (n = 11) had peritumoral edema on preoperative MRI. Nine patients (30%) presented preoperative hydrocephalus. Digital subtraction angiography was available in 4 patients (13%), and preoperative embolization was performed on 3 (10%). Clinically, 21 patients (70%) showed signs of cerebellar syndrome and 19 patients (63%) had intracranial hypertension; whereas, in another 10 cases (33%), intractable hiccups, lower cranial nerve deficit, and cervicalgia were diagnosed.
The most common tumor localization corresponded to the posterior and lateral regions of the cerebellar hemispheres. Surgery was performed using a midline or retromastoid suboccipital approach. The surgical procedure comprised peritumoral tumor dissection along the interphase between tumor adjacent parenquima, with coagulation of small afferent vessels to control bleeding, which can occur with central debulking. For cystic cavities, once contents had been drained, cyst walls were carefully checked for satellite nodules, whereas normal-appearing wall was spared. Complete surgical resection was accomplished in 28 cases (93%). In 2 patients (7%), resection was subtotal due to intraoperative bleeding. Four patients (13%) required intraoperative transfusions because of tumor bleeding. Two patients (6.6%) presented postoperative bleeding from the surgical site, but responded to medical management. Only 1 patient with preoperative hydrocephalus required external ventricular drainage during the postoperative period. Postoperative MRI scans confirmed subtotal resection in 2 patients and tumor recurrence in 3 patients (10%). Tumor recurrence cases included 2 VHL patients who developed new lesions and 1 who after complete resection developed local recurrence. Eighty percent (n = 4) of patients with tumor remnant or recurrence were reoperated after developing symptoms.
Complications observed included cerebrospinal fluid fistula (3.3%), pulmonary embolism (3.3%), meningitis (6.6%), urinary infection (3.3%), and cerebellar isquemia (3.3%). HBL diagnosis was confirmed by histology in all cases. Median duration of follow-up was 24 months (range: 3–18 months). Postoperative course according to the modified Raven scale (mRS) score showed good results in 25 patients (83%), fair in 3 (10%), and poor in 2 (7%).
Long-term follow-up indicated good clinical course in 28 patients (93.3%), intermediate in 1, and poor in 1 (3.3%) [
Regarding types of lesions observed, although more cystic HBLs were diagnosed, no statistical differences between cystic and solid tumors were observed in relation to immediate (Fisher’s exact test; P = 0.729), or long-term (Fisher’s exact test; P = 0.480) postoperative follow-up. This could in part be due to the limited sample size, as at least 44 cases per group would be required to detect a 20% difference between them with a power of 0.8.
Sample cases
Case 1. Sporadic cerebellar lesion
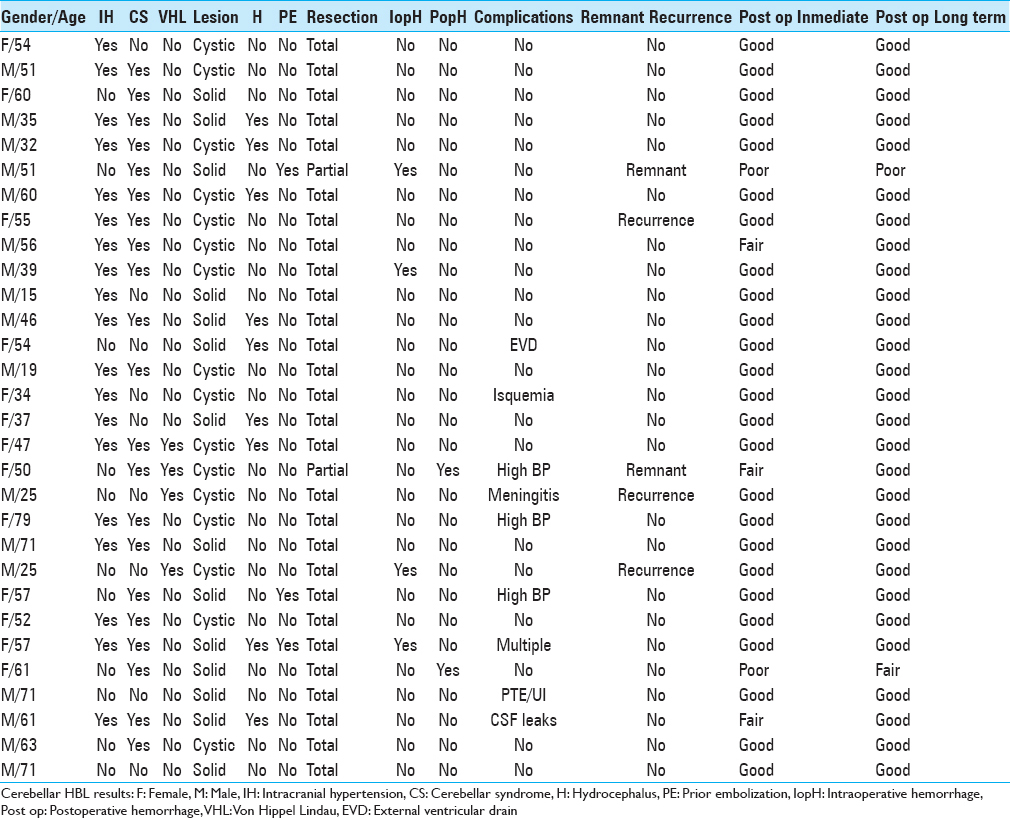
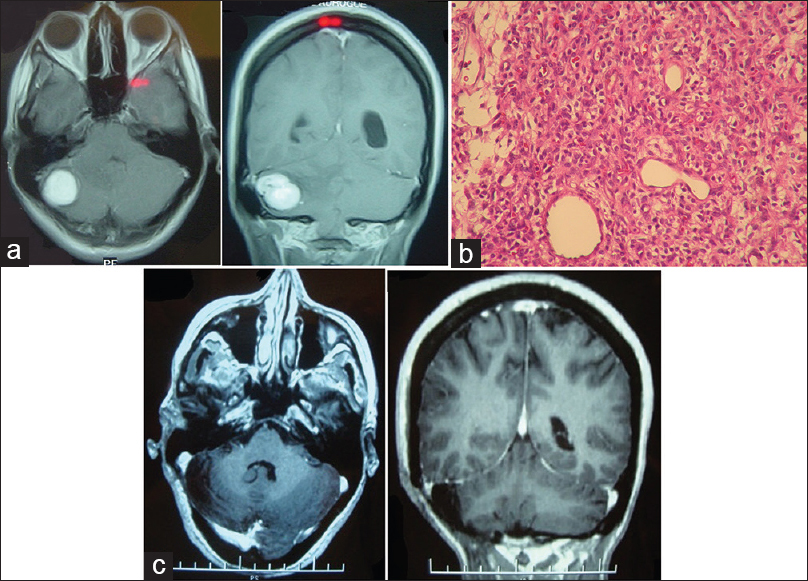
A 34-year-old female patient presented with recent-onset headaches in the occipitocervical region. On MRI, a solid lesion was observed in the right cerebellar hemisphere, with perilesional edema, marked contrast uptake, and compression of the fourth ventricle, but without hydrocephalus. Complete surgical excision was achieved through a suboccipital retromastoid approach. mRs score after surgery was 1. HBL microscopy specimen showed stromal cells (large, with vacuoles, and high lipid content) and numerous thin-walled vessels (shown on CD34 immunohistochemistry staining). Reticulin stain revealed a reticular pericellular network. Six years after surgery, no lesion recurrence has been detected and the mRS score remains 0 [
Case 2. Cerebellar lesion in a patient with von Hippen-Lindau disease
A 40-year-old-female patient with VHL disease and multiple retinal hemangioblastomas, causing amaurosis of the right eye, along with an asymptomatic left-sided cerebellar lesion, later developed progressive left-sided hearing loss and intracranial hypertension. On MRI, growth of the cystic component of the cerebellar lesion was observed, with compression of the IV ventricle and hydrocephalus. Preoperative mRS score was 1. No postoperative complications were observed after complete surgical resection of the lesion (postoperative mRS score was 0). No cerebellar lesion recurrence was found after 14 months of follow-up, although an asymptomatic cystic lesion was observed in the dorsal spine [
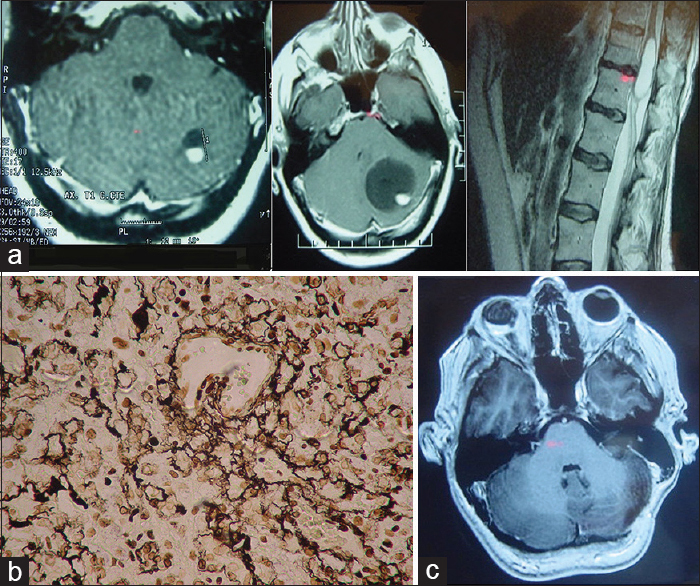
Figure 2
(a) Preoperative MRI of the brainstem and spine. T1-weighted axial section with contrast 1 year before surgery (left), at time of surgery (center) y and T2-weighted spinal sagittal section showing asymptomatic intramedullary cyst. (b) Tumor microscopy. Reticulin stain, magnification ×40. (c) Postoperative MRI showing complete resection
DISCUSSION
HBLs are vascularized, well-circumscribed, solid or cystic neoplasms of the CNS and are generally benign. They represent between 1 and 2.5% of all CNS tumors and are found predominantly in the cerebellum, less often in the brainstem or spinal cord, and very rarely in the supratentorial region or along nerve roots.[
Approximately 80% are sporadic, whereas 20–25% develop in the context of VHL disease.[
Magnetic resonance is not pathognomonic, and hence, the definitive diagnosis is made on histopathological examination. Microscopic investigation shows an extensive vascular network with neoplastic stromal cells. The neoplastic stromal cells have abundant cytoplasm with lipid vacuoles resulting in a typical clear cell morphology. Nuclear hyperchromatism and atypia, in contrast to the absence of mitotic activity, are a very typical pattern of HBLs.[
von Hippel–Lindau syndrome
In 1904, Eugene von Hippel, an ophthalmologist, reported finding retinal angiomatosis in 2 patients. However, it was Avrid Lindau, a pathologist, who later described the VHL syndrome, linking retinal angiomas to the presence of cerebellar lesions.[
VHL disease is transmitted in autosomal dominant fashion with incomplete penetrance and has a reported incidence of 1 per 36000 to 53000 live births. It is caused by the mutation of the VHL gene on the short arm of chromosome 3p25-6. Patients present with cerebellar (44–72%), spinal cord (40–50%), brainstem and retina (10–25%) HBLs, as well as neoplasms in other specific sites. Sixty to ninety percent of patients with VHL disease develop multiple and recurrent HBLs.[
In 2014, Lonser et al. reported a series of 225 patients with VHL disease, followed for almost 7 years. Tumors were located in the supratentorial compartment (1%), cerebellum (45%), brainstem (7%), spinal cord (36%), cauda equina (11%), and in nerve roots (0.3%), with, on average, 7 lesions per patient detected at the time of study entry. By the end of the follow-up, 72% patients had developed new lesions (predominantly in the cerebellum and spinal cord). HBLs increased in volume on average 3.7 mm3/year, observed most often in cases of cerebellar and spinal cord neoplasms, as well as in male patients.[
Lonser described three types of tumor growth pattern – saltatory (72%), linear (6%), and exponential (22%).[
In the Lonser study, 93.6% of HBLs remained asymptomatic during follow-up, whereas 6.3% developed secondary symptoms resulting from mass effect caused by either the lesion and/or the cyst.[
The leading cause of death was HBL progression in 25% of the cases studied, followed by renal cell carcinoma (RCC).[
Other syndrome findings include:
Retinal or optic nerve HBLs. Present in 60% of the cases, these may be multifocal or bilateral. Bleeding causes blindness, retinal detachment, and glaucoma. Treatment options include laser photocoagulation or cryotherapy, although larger lesions may require enucleation of the eye.[ Adrenal or extra-adrenal pheocromocytomas develop in 10% of the cases, and may require surgical resection in symptomatic cases Pancreatic cysts, serous cystadenomas, and neurendocrine tumors are present in 70% of the cases Papillary cystadenoma of the middle ear, an endolymphatic sac tumor, has been reported in 15% of the patients. This tumor may cause hearing loss, vertigo, and facial palsy. Treatment is surgical excision.
Currently, three variants of VHL syndrome have been identified:
Type 1: High risk of clear cell RCC, but not of pheochromocytoma Type 2a: High risk of pheochromocytoma, low risk for RCC Type 2b: Risk of pheochromocytoma and RCC Type 2c: Risk of pheochromocytoma only Type 3: Chuvash polycythemia without tumor lesions.
VHL syndrome diagnosis requires both clinical criteria, and more recently, detection of genetic mutation in peripheral blood.[
Diagnostic criteria include:
Presence of typical VHL lesion in a patient with family history of VHL, or Patients with family history of VHL, two or more HBLs in the CNS, or a single HBL lesion in combination with other typical VHL lesions.
MRI with contrast should be used to study sporadic craniospinal lesions. Also, ophthalmoscopic exam, abdominal ultrasound, audiometry, and vanillin-mandelic acid (VMA) assay performed on a 24-hour urine sample to rule out VHL are useful. If the latter test is positive, genetic counseling for first-degree relatives is recommended.[
Patient follow-up should be long-term for early detection of new HBLs, kidney tumors, and retinal angiomas, and includes:
Yearly: (1) Clinical assessment of urinary tract function, (2) VMA assay performed on a 24-hour urine sample, (3) ophthalmoscopic exam, and (4) renal ultrasound Every 3 years: (q) MRI of the entire CNS, with and without contrast, (2) audiometry, (3) VMA assay performed on a 24-hour urine sample, and (4) abdominal CT scan with and without contrast.[
Treatment of choice is the surgical resection of symptomatic lesions or those presenting growth on imaging. Asymptomatic lesions may be followed using clinical and imaging methods.[
Cerebellar hemangioblastomas
The most common localization of HBLs is the cerebellum, both for sporadic forms or for those associated with VHL.[
As published in the literature, in this series, patients presenting HBL in the context of VHL disease first developed symptoms at an earlier age than sporadic cases.[
Cytology of cyst fluid showed stromal cells with round or oval nuclei, eosinophilic foamy cytoplasm with vacuoles, as well as endothelial cells and some macrophages[

The main differential diagnoses are metastasis and astrocytomas.[
Surgical treatment is recommended for symptomatic lesions or those increasing in size on imaging. Large, deep, or highly vascularized lesions may benefit from preoperative embolization with n-butyl cyanoacrylate or onyx 18.[
Hojo et al. reported in 2014 the use of indocyanine green to distinguish afferent tumor vessels from “en passage” vascular structures, as well as to confirm complete excision of the tumor nodule.[
Ninety-four percent of hydrocephalus-associated cases resolved after HBL resection.[
Ten to twenty percent of cerebellar HBLs linked to VHL disease also presented reactive polycythemia, secondary to the secretion of erythropoietin or an erythropoietin-like substance secreted by the tumor nodule, which in general, disappeared after HBL resection.[
Functional postoperative results vary between series and are worse for patients with solid lesions, probably because of greater perilesional edema.[
Postoperative tumor recurrence ranged 17–75% for patients with VHL and 5–17% for sporadic cases.[
Postoperative bleeding has been reported to be 3–16%, and the most common complications include infections (3.15%) and cerebellar isquemia (2.85%).[
Recurrence
Postoperative tumor recurrences are more common in males, with symptom onset prior to 30 years, presenting multiple, small, solid lesions, with high stromal eosinophil cell count.[
Hemangioblastomatosis is a rare complication in VHL patients characterized by diffuse leptomeningeal tumor spread. Antiangiogenic treatment has been proposed for these cases using agents such as interferon alfa-2a, vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor SU5416, temozolamide receptor inhibitor, bevacizumab, and erlotinib. These treatments will in general produce rapid subjective improvement without radiological changes.[
Radiosurgery
In the last 25 years, stereotactic radiosurgery has been used as the first line treatment or as adjuvant or salvage therapy in some HEB patients. Although use is still debated, it may be considered for selected cases; for example, patients with prior tumor growth may achieve tumor control with radiosurgery in 79–92% of the cases.[
In a series reported by Asthagiri et al., 42 posterior fossa HEBs were treated with radiosurgery. Gamma Knife dose was 18.1 Gy (range: 12–20 Gy), with a maximum radiation dose of 34.6 Gy (range: 23–49 gy) along tumor margins and a 54% mean isodose line (range: 35–85%). Dose administered in LINAC treatment plan was 25.1 Gy (range: 15–34 Gy), and 20 Gy (range: 12–24 gy) along tumor margins with 82% mean isodose line (range 70-965 GY). Progression-free survival at 2 years was 91%, decreasing to 61% and 51% at 10 and 15 years follow-up, respectively. Edema or cystic degeneration as well as small-sized lesion progression on imaging led authors to suggest the limiting the role of radiosurgery to lesions in which surgical resection may imply high risk sequelae for patients.[
In 2014, Puataweepong et al. reported results for radiosurgery and LINAC-based fractionated radiotherapy. Small lesions (<3 cm) were treated in a single session with a mean dose of 20 Gy. Lesions greater than 3 cm in the brainstem or the optic nerves received one session of <10–12 Gy, or a series of 2–5 sessions of fractionated radiotherapy, if tolerance to radiation to these structures was surpassed. However, results obtained did not differ significantly from previously published findings, with global tumor control rates of 98%, 88%, and 73% at 1, 2, and 6 years, respectively.
In agreement with other series, 2 of 14 patients treated required emergency surgery because of cystic component growth, due to which authors do not recommend radiation of predominantly cystic lesions.[
CONCLUSIONS
Symptoms generated by cerebellar HBLs depend on the presence of edema, tumor-associated cysts, and lesion growth rate. Surgical resection is the treatment of choice for sporadic HBLs, those associated with VHL disease, or those showing growth on imaging. The surgical technique involves circumferential lesion dissection, always attempting en-bloc resection to avoid intraoperative bleeding. For small, solid, difficult to access lesions, radiosurgery ablation is an option. Small asymptomatic lesions in patients with VHL disease should be followed closely long-term, as delayed lesion growth may occur. All cases of HBL should be screened for the VHL gene mutation; if the test is positive, genetic counseling for family members at risk is warranted.
Study limitations
This is a retrospective series, which is a limitation of the study. Sporadic cases were reviewed together with cases linked to VHL disease, although long-term prognosis is less favorable in VHL cases. True prevalence of VHL may have been underestimated as not all patients completed full evaluation to rule out or confirm the diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Akimoto J, Fukuhara H, Suda T, Nagai K, Hashimoto R, Michihiro K. Disseminated cerebellar hemangioblastoma in two patients without von Hippel-Lindau disease. Surg Neurol Int. 2014. 5: 145-54
2. Ammerman JM, Lonser RR, Dambrosia J, Butman JA, Oldfield EH. Long-termnatural history of hemangioblastomas in patients with von Hipel-Lindau disease: Implications for treatment. J Neurosurg. 2006. 105: 248-55
3. Asthagiri AR, Mehta GU, Zach L, Li X, Butman JA, Camphausen KA. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro-Oncology. 2010. 12: 80-6
4. Bamps S, van Calenbergh F, de Vleeschouwer S, Loon JV, Sciot R, Legius E. What the neurosurgeon should know about hemangioblastoma, both sporadic and in Von Hippel-Lindau disease: A literature review. Surg Neurol Int. 2013. 4: 145-54
5. Bishop FS, Liu JK, Chin SS, Fults DW. Recurrent cerebellar hemangioblastoma with enhancing tumor in the cyst wall: Case report. Neurosurgery. 2008. 62: E1378-89
6. Brundl E, Schodel P, Ullrich OW, Brawanski A, Schebesch KM. Surgical resection of sporadic and hereditary hemangioblastoma: Our 10-year experience and a literature review. Surg Neurol Int. 2014. 5: 138-42
7. Bush ML, Pritchett C, Packer M, Ray-Chaudhury A, Jacob A. Hemangioblastoma of the cerebellopontine angle. Arch Otolaryngol Head Neck Surg. 2010. 136: 734-8
8. Byung HM, Sang Kyu P, Young-Min H. Large Solid Hemangioblastoma in the Cerebellopontine Angle: Complete Resection Using the Transcondylar Fossa Approach. Brain Tumor Res Treat. 2014. 2: 128-33
9. Campero A, Ajler P, Fernandez J, Isolan G, Paiz M, Rivadeneira C. Hemangioblastoma of the posterior fossa: Report of 16 cases and review of the literature. Surg Neurol Int. 2016. 7: 855-60
10. Conway JE, Chou D, Clatterbuck RE, Brem H, Long DM, Rigamonti D. Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery. 2001. 48: 55-63
11. Da Costa LB, de Andrade A, Braga BP, Ribeiro CA. Cauda equina hemangioblastoma. Case report. Arq Neuropsiquiatr. 2003. 61: 456-8
12. De La Monte SM, Horowitz SA. Hemangioblastomas. Clinical and histopathological factors correlated with recurrence. Neurosurgery. 1989. 25: 695-8
13. Epari S, Bhatkar R, Moyaidi A, Shetty P, Gupta T, Kane S. Histomorphological spectrum and immunohistochemical characterization of hemangioblastomas: An entity of unclear histogenesis. Indian J Pathol Microbiol. 2014. 57: 542-8
14. Fukuda M, Takao T, Hiraishi T, Yoshimura J, Yajima N, Saito A. Clinical factors predicting outcomes after surgical resection for sporadic cerebellar hemangioblastomas. World Neurosurg. 2014. 82: 815-21
15. Hes FJ, Feklberg MA. von Hippel-Lindau disease: Strategies in early detection (renal, adrenal, pancreatic masses). Eur Radiol. 1999. 9: 598-610
16. Hojo M, Arakawa Y, Funaki T, Yoshida K, Kikuchi T, Takagi Y. Usefulness of tumor blood flow imaging by intraoperative indocyanine green videoangiography in hemangioblastoma surgery. World Neurosurg. 2014. 82: 495-501
17. Horvathy DB, Hauck EF, Ogilvy CS, Hopkins LN, Levy EI, Siddiqui AH. Complete preoperative embolization of hemangioblastoma vessels with Onyx 18. J Clin Neurosci. 2011. 18: 401-3
18. Jagannathan J, Lonser RR, Smith R, DeVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2008. 108: 210-22
19. Koh ES, Nichol A, Millar BA, Ménard C, Pond G, Laperriere NJ. Role of fractionated external beam radiotherapy in hemangioblastomas of the central nervous system. Int J Radiat Oncol Biol Phys. 2007. 69: 1521-6
20. Law EKC, Lee RKL, Griffith JF, Siu DY, Ng HK. Spinal nerve root haemangioblastoma associated with reactive polycythemia. Case Rep Radiol. 2014. 2014: 798620-
21. Lallu S, Naran S, Palmer D, Bethwaite P. Cyst fluid cytology of cerebellar hemangioblastoma; a case report. Diagn Cytopathol. 2008. 36: 341-3
22. Lee GJ, Jung TY, Kim IY, Jung S, Jang WY, Moon KS. The clinical experience of recurrent central nervous system hemangioblastomas. Clin Neurol Neurosurg. 2014. 123: 90-5
23. Lonser RR, Butman JA, Huntoon K, Asthagiri AR, Wu T, Bakhtian KD. Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg. 2014. 120: 1055-62
24. Lonser RR, Butman JA, Kiringoda R, Song D, Oldfield EH. Pituitary stalk hemangioblastomas in von Hippel-Lindau disease: Clinical article. J Neurosurg. 2009. 110: 350-3
25. Lonser RR, Vortmeyer AO, Butman JA, Glasker S, Finn MA, Ammerman JM. Edema is a precursor to central nervous system peritumoral cyst formation. Ann Neurol. 2005. 58: 392-9
26. Moss JM, Choi CY, Adler JR, Soltys SG, Gibbs IC, Chang SD. Stereotactic radiosurgical treatment of cranial and spinal hemangioblastomas. Neurosurgery. 2009. 65: 79-85
27. Neumann HPH, Eggert HR, Weigel K, Friedburg H, Wiestler OD, Schollmeyer P. Hemangioblastomas of the central nervous system. A 10 year study with special reference to von Hippel-Lindau syndrome. J Neurosurg. 1989. 70: 24-30
28. Omar A. Bevacizumab for the treatment of surgically unresectable cervical cord hemangioblastoma: A case report. J Med Case Rep. 2012. 6: 238-
29. Padhi S, Sarangi R, Challa S, Bussary P, Panigrahi MK, Purohit AK. A 10-year retrospective study oh hemangioblastomas of the central nervous system with reference to von Hippel-Lindau (VHL) disease. J Clin Neurosci. 2011. 18: 939-44
30. Peyre M, David P, van Efenterre R, François P, Thys M, Emery E. Natural history of supratentorial hemangioblastomas in von Hippel-Lindau disease. Neurosurgery. 2010. 67: 577-87
31. Puataweepong P, Dhanachai M, Hansasuta A, Dangprasert S, Sitathanee C, Puddhikarant P. The clinical outcome of intracranial hemangioblastomas treated with linac-based stereotactic radiosurgery and radiotherapy. J Radiat Res. 2014. 55: 761-8
32. Rachinger J, Buslei R, Prell J, Strauss C. Solid hemangioblastomas of the CNS: A review of 17 consecutives cases. Neurosurg Rev. 2009. 32: 37-48
33. Richard S, Campello S, Taillandier L, Parker F, Resche F. Hemangioblastoma of the central nervous system in von Hippel-Lindau disease. J Intern Med. 1998. 243: 547-53
34. Riklin C, Seystahl K, Hofer S, Happold C, Winterhalder R, Weller M. Antiangiogenic treatment for multiple CNS hemangioblastomas. Onkologie. 2012. 35: 443-5
35. Rogers LR, LoRusso P, Nadler P, Malik G, Shields A, Kaelin W. Erlotinib therapy for central nervous system hemangioblastomatosis associated with von Hippel–Lindau disease: A case report. J Neurooncol. 2011. 101: 307-10
36. Sakamoto N, Ishikawa E, Nakai Y, Akutsu H, Yamamoto T, Nakai K. Preoperative endovascular embolization for hemangioblastoma in the posterior fossa. Neurol Med Chir (Tokyo). 2012. 52: 878-84
37. Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel–Lindau Disease: Molecular Pathological Basis, Clinical Criteria, Genetic Testing, Clinical Features of Tumors and Treatment. Jpn J Clin Oncol. 2006. 36: 337-43
38. Vortmeyer AO, Falke EA, Glasker S, Li J, Oldfield EH. Nervous system involvement in von Hippel-Lindau disease: Pathology and mechanisms. Acta Neuropathol. 2013. 125: 333-50
39. Wan J, Chui H, Wang Y. Surgical management of large solid hemangioblastomas of the posterior fossa. J Clin Neurosci. 2011. 18: 39-42
40. Wang C, Zhang J, Liu A, Sun B. Surgical management of medullary hemangioblastoma. Report of 47 cases. Surg Neurol. 2001. 56: 218-26
41. Wang EM, Pau L, Wang BJ, Zhang N, Zhou LF, Dong YF. The long-term results of gamma knife radiosurgery for the hemangioblastomas of the brain. J Neurosurg (Suppl). 2005. 102: 225-9
42. Ye DY, Bakhtian KD, Asthagiri AR, Lonser RR. Effect of pregnancy on hemangioblastoma development and progression in von Hippel-Lindau disease. J Neurosurg. 2012. 117: 818-24

