

- Department of Neurology, Hanoi Medical University Hospital, Hanoi Medical University, Hanoi, Vietnam.
- Institute of Theoretical and Applied Research, Duy Tan University, Da Nang, Vietnam.
- Department of General Internal Medicine, Vinmec International Hospital, Hanoi, Vietnam.
DOI:10.25259/SNI_489_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Huong Van Nguyen1, Diep Ngoc Nguyen2, Huong Thi Nguyen3. A case of Wilson disease with the ATP7B mutation presenting movement disorders. 28-Jun-2021;12:303
How to cite this URL: Huong Van Nguyen1, Diep Ngoc Nguyen2, Huong Thi Nguyen3. A case of Wilson disease with the ATP7B mutation presenting movement disorders. 28-Jun-2021;12:303. Available from: https://surgicalneurologyint.com/surgicalint-articles/10933/
Date of Submission
17-May-2021
Date of Acceptance
27-May-2021
Date of Web Publication
28-Jun-2021
Abstract
Background: Wilson disease is an autosomal recessive condition manifested when abnormal copper accumulation in the body particularly involving many organs such as brain, liver, and cornea. Diagnosis is challenging with the completion of tests in blood and urine, a liver biopsy, and clinical evaluation. ATP7B mutations with more than 600 identified variants are the genetic disorders of Wilson disease.
Case Description: We report an adolescent case with no family history presented with extrapyramidal dyskinesia. Other symptoms include liver cirrhosis and Kayser–Fleischer ring. The typical presentation of blood test results and brain MRI images helps us to suspect Wilson disease in this case. We confirmed to have a p.R778L form and a p.S105X form in ATP7B mutations. After combining therapy with trihexyphenidyl and trientine, the patient’s medical condition was stable and no side effects were observed.
Conclusion: Screening for the diagnosis of Wilson disease is essential in helping patients benefit from early treatment and genetic counseling.
Keywords: ATP7B, Movement disorders, p.R778L, p.S105X, Wilson disease
INTRODUCTION
Wilson disease is an autosomal recessive condition occasionally caused by the ATP7B mutation, which results in the collection of superfluous copper in the liver, brain, cornea, and other organs. The prevalence of this disease is approximately 1/30,000 persons, both males and females are affected in the same way.[
CASE REPORT
Clinical features
A 17-year-old girl presented with hand tremors at rest and worsened by stress. There was no family history of hereditary disease and metabolic disorders. Tracing back her medical history, it was found that the onset of the tremors in both hands had occurred for 6 months. The patient’s handwriting was atrocious and illegible. Her daily activities and learning became more problematic and difficult. Clinical examination revealed the features of extrapyramidal symptoms included tremors with a frequency of about 4–6 Hz, mainly in the upper extremities, slow and scribbled writing. Furthermore, there were no signs of pyramidal tract lesions, ophthalmoplegia or hallucinations, and behavior disorders. Although the patient had malaise, she could still manage to walk independently. The clinical features of slowly progressing extrapyramidal symptoms in a young patient suspected that a metabolic disorder or intoxication affecting central gray nuclei.
With respect to laboratory findings, that diagnosis was supported. The elevated 24 h ceruloplasmin level (224 μg/day [<40 μg/day]) and the decreased serum ceruloplasmin level (3 mg/dL [20–40 mg/dL]) were identified. Complete blood count biochemistry and urinary routine were within normal limits. Brain MRI showed high signal intensities of both basal ganglia, head of caudate nucleus in T2W, fluid-attenuated inversion recovery, and diffuse-weighted imaging [
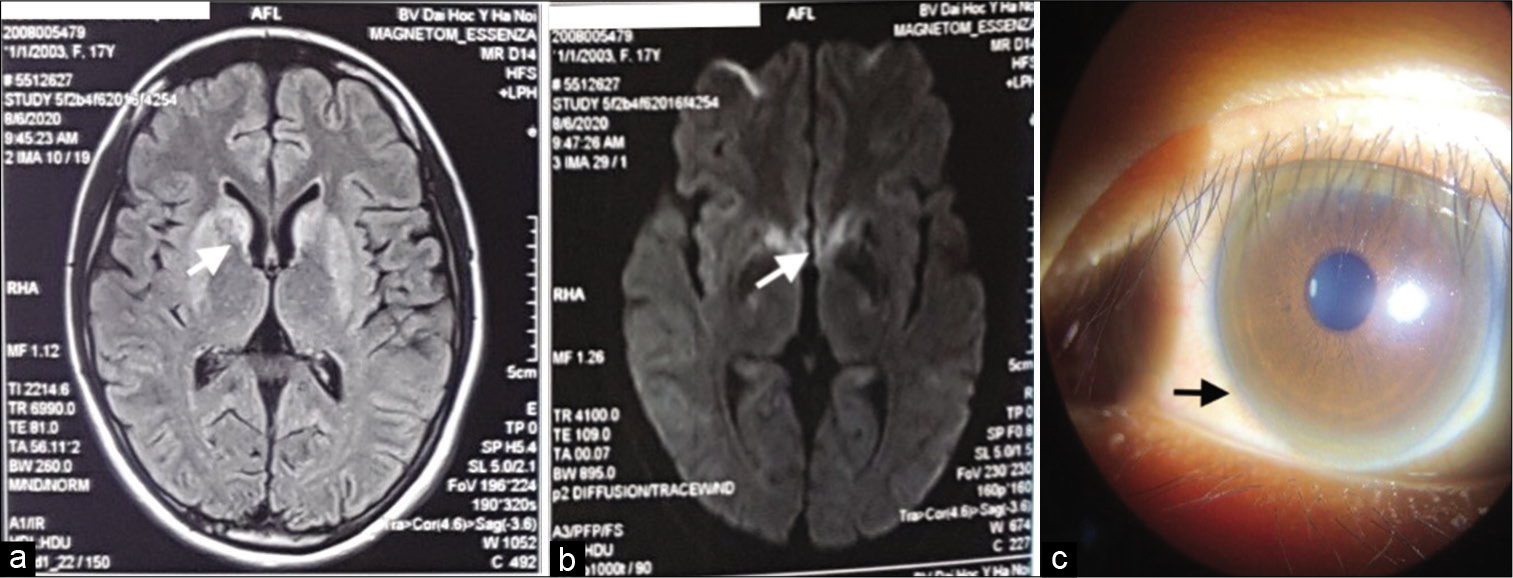
Treatment results
The patient has received trihexyphenidyl (10 mg/day) and trientine (1.5 g/day) and had no significant side effects. After about 6 months of treatment, the movement function has partially improved, her writing and movement have been more flexible.
DISCUSSION
Wilson disease has been described since 1912 as a copper metabolic disorder, with neurological manifestations, cirrhosis and eye symptoms caused by superfluous copper collection in many organs.[
The initial symptoms of the disease in approximately 50% of patients are neurologic. Basal ganglia-based movement disorders are features of the disease. Asymmetric tremor is the first neurologic characteristic in approximately 50% of individuals and may be proximal or distal and manifest at rest or with movement. Other presentations include dysarthria, dysphasia, ataxia, and Parkinsonian-like extrapyramidal signs. In children, early signs are fine motor disorders as atrocious and illegible handwriting. Later, patients present with dystonia, spastic quadriplegia, and possible seizures. Clinical examination usually reveals generalized extrapyramidal hypertonia. Mood disorders are common, but depression and behavioral disturbances can occur. Kayser–Fleisher rings are the result of the deposition of copper in the descemet membrane of the cornea. These findings with the rings are usually enough for diagnosis but the most accurate test for an alternative diagnosis is a liver biopsy.
About 95% of Wilson patients have serum ceruloplasmin below 20 mg/dl (normal range 20–40 mg/dl). Meanwhile, increasing the urine ceruloplasmin over 100 μg/24 h (normal range <40 μg/24 h) is valid for screening and a diagnostic criterion for determining the disease. Wilson disease should be suspected if symptoms persist with the disease are present or if a proportionate has been found to determine the disease. Most had lightly abnormal liver function tests and increased aspartate transaminase, alanine transaminase, and bilirubin levels. If damaging the liver significantly, the prothrombin time is prolonged since clotting factors are not produced enough. If neurological symptoms are present, brain MRI may show hyperintensities in the basal ganglia in the T2 sequence.
The first established diagnosis criteria were Sternlieb (1978), then Ferenci (2003) added genetic analysis factors for early diagnosis.[
The treatment for Wilson disease consists of copper chelation therapy, reducing the symptoms, and physiotherapy. The mainstay chelation is preferred is trientine because of its side effects. Anticholinergics (trihexyphenidyl), GABA antagonists, and levodopa can be used for muscle rigidity and Parkinsonian features. The benefits of liver transplantation in improving neurological disorders in some patients responding inadequately to basic therapy were demonstrated.[
Our patient had an adolescent onset with inconspicuous neurological manifestations, characterized by bilateral extrapyramidal syndrome with hand tremors and writing disorders, cirrhosis, and the typical eye sign. The patient fully met diagnostic criteria with clinical N1 form (nerve combined with liver damage). Confirmed the diagnosis by genetic sequencing of the patient found two heterozygous mutations in the ATP7B gene, which are the stop codon mutation c.314C>A (p.S105X) and the missense mutation c. 2333G>T (p.R778L). In the c.314C>A mutation, the nucleotide sequence C is replaced to A, so the 105th TCG codon encodes serine (S) to replace it into the TAG (X) terminating. In the c.2333G> T mutation, the nucleotide sequence G replaced T resulted in a 778th CGG encoding arginine (R) to convert to leucine (L).
According to the previous studies, patients with the p.S105X mutation often had clinically severe signs caused by the complete loss of copper transport function of ATP7B protein. The more mutant allele appear, the more the copper transportation of the ATP7B protein is affected, which correlates with the earlier and more severe clinical manifestations. Our patient exhibits hepatic neurological coordination with poor clinical symptoms but severe subclinical manifestations with exceptionally low urinary ceruloplasmin concentration, eye damage, and cirrhosis.
We recommend analyzing two maker mutation regions in the ATP genes of a patient’s family members, especially to determine which member carries the p.S105X mutation (a missense mutation that can cause symptoms even carrying one mutant allele) for early diagnosis and treatment without clinical manifestations.
CONCLUSION
Wilson disease is a rare genetic condition caused by a metabolic disorder that causes copper deposits in many organs. Neurological manifestations are usually inconspicuous, mostly following liver damage. Our clinical case with early onset of juvenile extrapyramidal syndrome suggests screening for liver and eye lesions. In addition to conducting genetic sequencing to identified mutations to provide an important basis for genetic counseling, patients can benefit from early treatment. It is important to keep in mind the genetic etiology in a young patient presenting with extrapyramidal symptoms even with no relevant family history.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Dmitriev OY, Bhattacharjee A, Nokhrin S, Uhlemann EM, Lutsenko S. Difference in stability of the N-domain underlies distinct intracellular properties of the E1064A and H1069Q mutants of copper-transporting ATPase ATP7B. J Biol Chem. 2011. 286: 16355-62
2. Ferenci P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: Impact on genetic testing. Hum Genet. 2006. 120: 151-9
3. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003. 23: 139-42
4. Poujois A, Sobesky R, Meissner WG, Brunet AS, Broussolle E, Laurencin C. Liver transplantation as a rescue therapy for severe neurologic forms of Wilson disease. Neurology. 2020. 94: e2189-202
5. Rosencrantz R, Schilsky M. Wilson disease: Pathogenesis and clinical considerations in diagnosis and treatment. Semin Liver Dis. 2011. 31: 245-59
6. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014. 133: 1-9
7. Sternlieb I. Diagnosis of Wilson’s disease. Gastroenterology. 1978. 74: 787-9
8. Thomas GR, Roberts EA, Walshe JM, Cox DW. Haplotypes and mutations in Wilson disease. Am J Hum Genet. 1995. 56: 1315-9
9. Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015. 16: 6419-31

