

- Department of Neurosurgery, Keio University, Shinanomachi, Shinjyukuku, Tokyo, Japan.
DOI:10.25259/SNI_211_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Nobuhiko Arai, Takenori Akiyama. A questionnaire-based survey to evaluate and improve the current HHT medical and social condition in Japan. 02-Oct-2020;11:323
How to cite this URL: Nobuhiko Arai, Takenori Akiyama. A questionnaire-based survey to evaluate and improve the current HHT medical and social condition in Japan. 02-Oct-2020;11:323. Available from: https://surgicalneurologyint.com/surgicalint-articles/10299/
Date of Submission
24-Apr-2020
Date of Acceptance
03-Aug-2020
Date of Web Publication
02-Oct-2020
Abstract
Background: Hereditary hemorrhagic telangiectasia (HHT) is a genetic systemic vascular disease affecting multiple organs and shows recurrent intractable symptoms. This disease has not been widely recognized in Japan until recently. Both diagnosed HHT patients and potential ones have faced difficulties because of the unfamiliarity with the disease in Japan. To evaluate the effect and degree of such a Japanese situation, a questionnaire-based survey was executed in this study.
Methods: This survey was carried out among the members of HHT Japan Association. The organization consisted of 102 members (as of 6/2019), mainly HHT patients and their family members. A questionnaire was used to gather demographic data, the effort to reach the diagnosis, and information regarding current patients’ and their families’ medical managements.
Results: Of the 102 questionnaires distributed, we have got 56 responses. The participants were mostly female (30) with an average age of 55.4 ± 14.8 (mean ± standard deviation [SD]) years. The average age of males was 53.5 ± 16.4. Relatively many HHT patients were born in huge cities such as Tokyo, Osaka, and Fukuoka Prefecture (n = 4 to 8 patients). The duration between the initial symptoms and the definite diagnosis was 8.8 ± 10.9 years. The number of hospitals involved in the final diagnosis was 2.38 ± 1.83. More than 70% of patients now have to visit at least two departments and 24% of HHT patients did not want their family to screen for HHT.
Conclusion: HHT medical practice in Japan should be further modified, for example, by establishing HHT centers and educating primary care physicians and HHT patients.
Keywords: Epistaxis, Hereditary hemorrhagic telangiectasia, Osler–Weber–Rendu disease, Quality of life
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease, is a genetic systemic vascular disease characterized by prominently recurrent epistaxis, cutaneous spot (telangiectasia), and arteriovenous shunt lesions in multiple organs.[
In addition to the problem of potential HHT patients, diagnosed HHT patients also suffer from improper medical management due to disease’s rarity and multisymptomatic features. HHT expresses frequent intractable epistaxis and affects multiple organs, which force the patient to visit multiple departments in different medical institutes. There are few literatures or surveys which demonstrated overall and detailed situations or demands of Japanese HHT patients. Through this study, we planned to evaluate the difficulty of HHT diagnosis, current situation of HHT patients, and the possibility to detect potential undiagnosed patient to combat the unrefined HHT clinical condition in Japan.
MATERIALS AND METHODS
This questionnaire-based survey was approved by the ethical committee of our hospital (No. 20190176) and distributed to the members of HHT Japan Association, a nonprofit organization founded in 2012, consisting of 102 members (as of 6/2019). The content of the questionnaire is shown in [

RESULTS
Background
Out of the 102 questionnaires distributed among the patients, 56 responses were recorded, and females were found to be dominant (30). The average age of males was 53.5 ± 16.4 (mean ± standard deviation [SD]) and females was 55.4 ± 14.8 (mean ± SD). There were more HHT patients from large cities in Japan such as Tokyo, Osaka, and Fukuoka Prefecture (n = 4–8 patients). The other patients are distributed across Japan.
Difficulty of diagnosis
The most frequent symptom which compelled the patients to visit the hospital was epistaxis (50% of HHT patients) and the second most frequent one was neurological deficit [
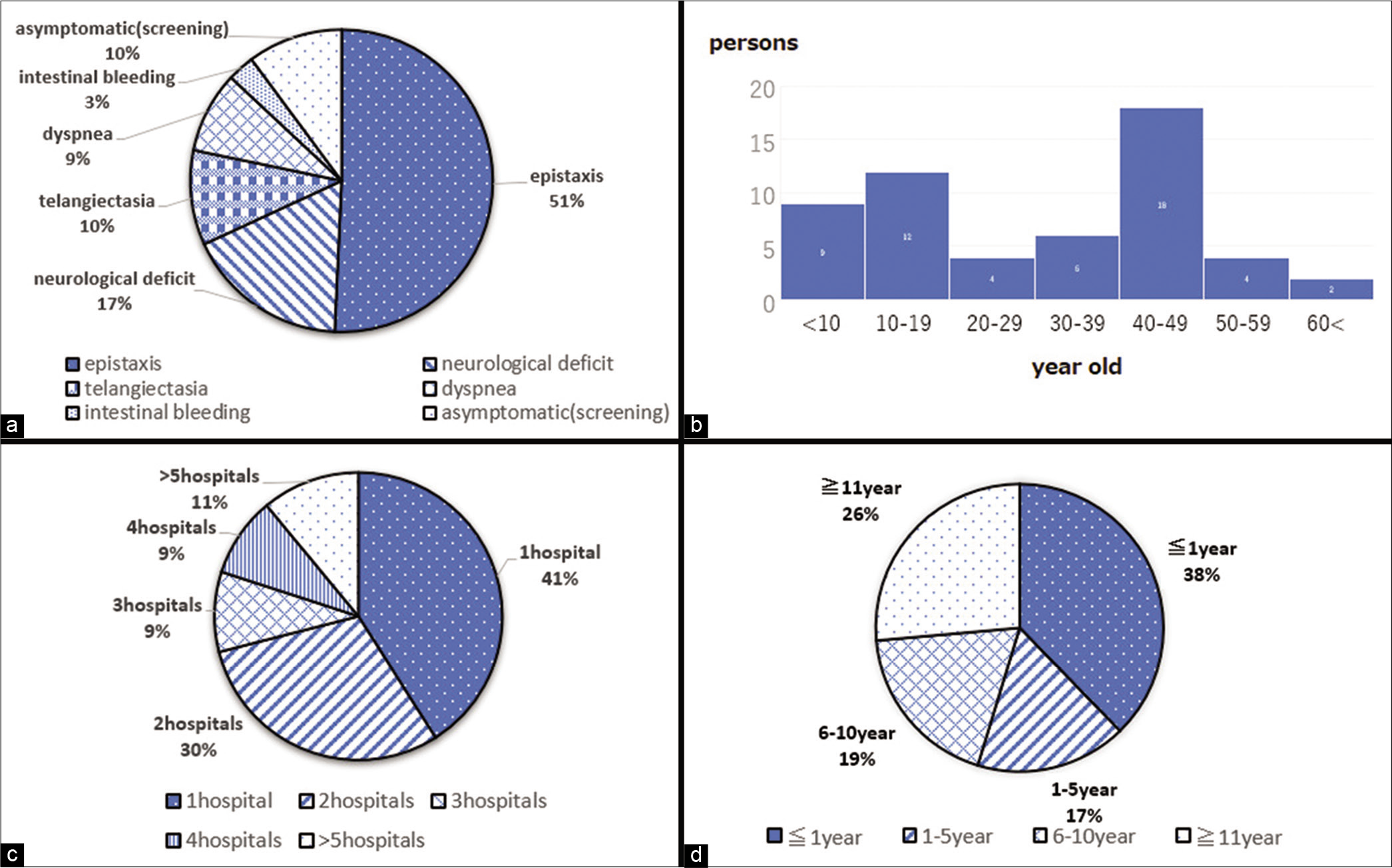
Current situation of HHT patients
As for the current frequency of nasal epistaxis, 45% of HHT patients suffer from nasal bleeding every day. As much as, 90% of patients had epistaxis at least once a week [
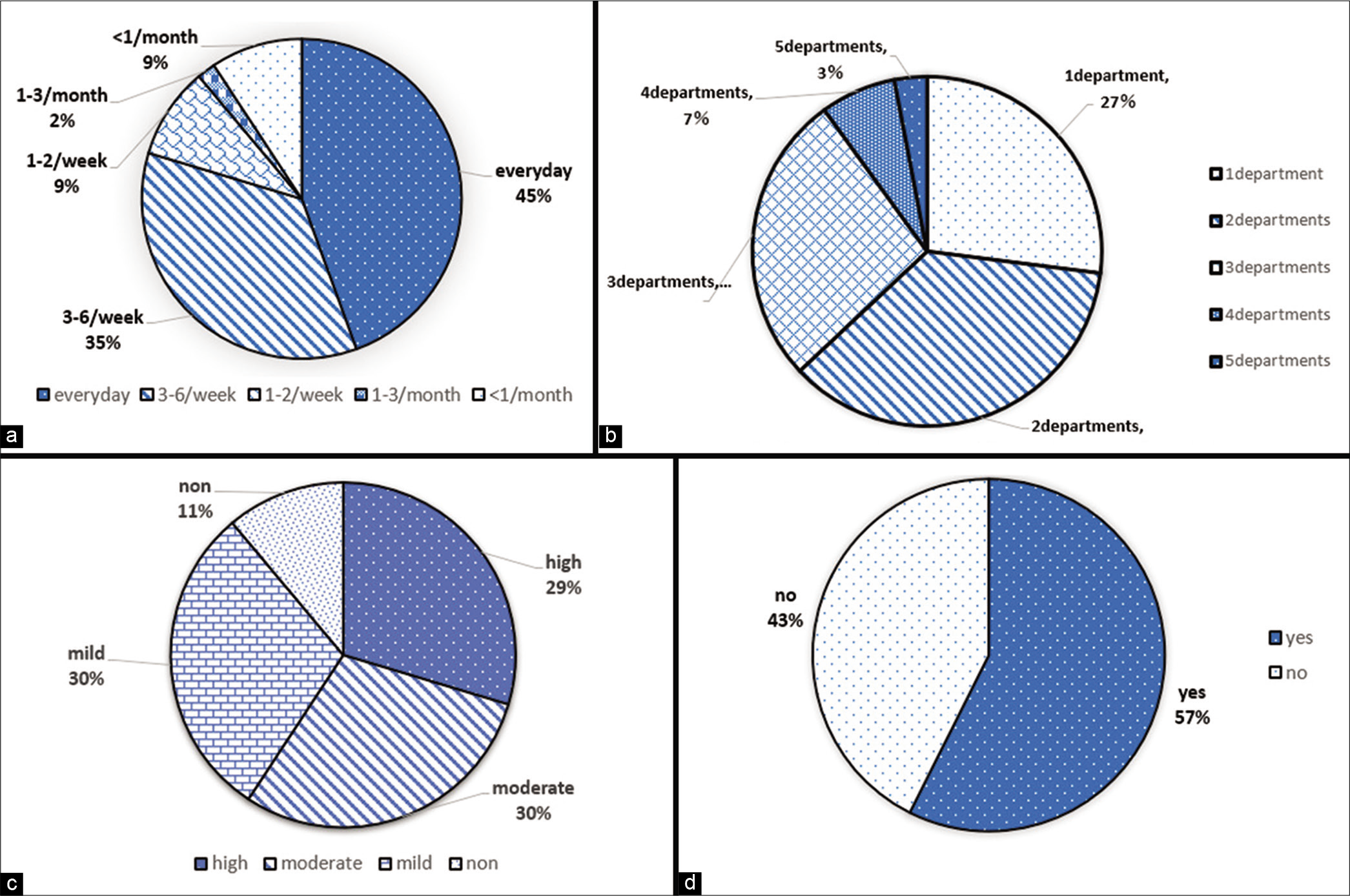
Figure 3:
Current data of hereditary hemorrhagic telangiectasia (HHT) patients. (a) Frequency of epistaxis. (b) Number of departments patients has to visit. (c) Feeling of economical oppression among patients. (d) Proportion of HHT patients who are supported by Ministry of Health, Labour and Welfare.
Potential HHT patients
Frequent nasal bleeding was noticed by family members for 82% of HHT patients [
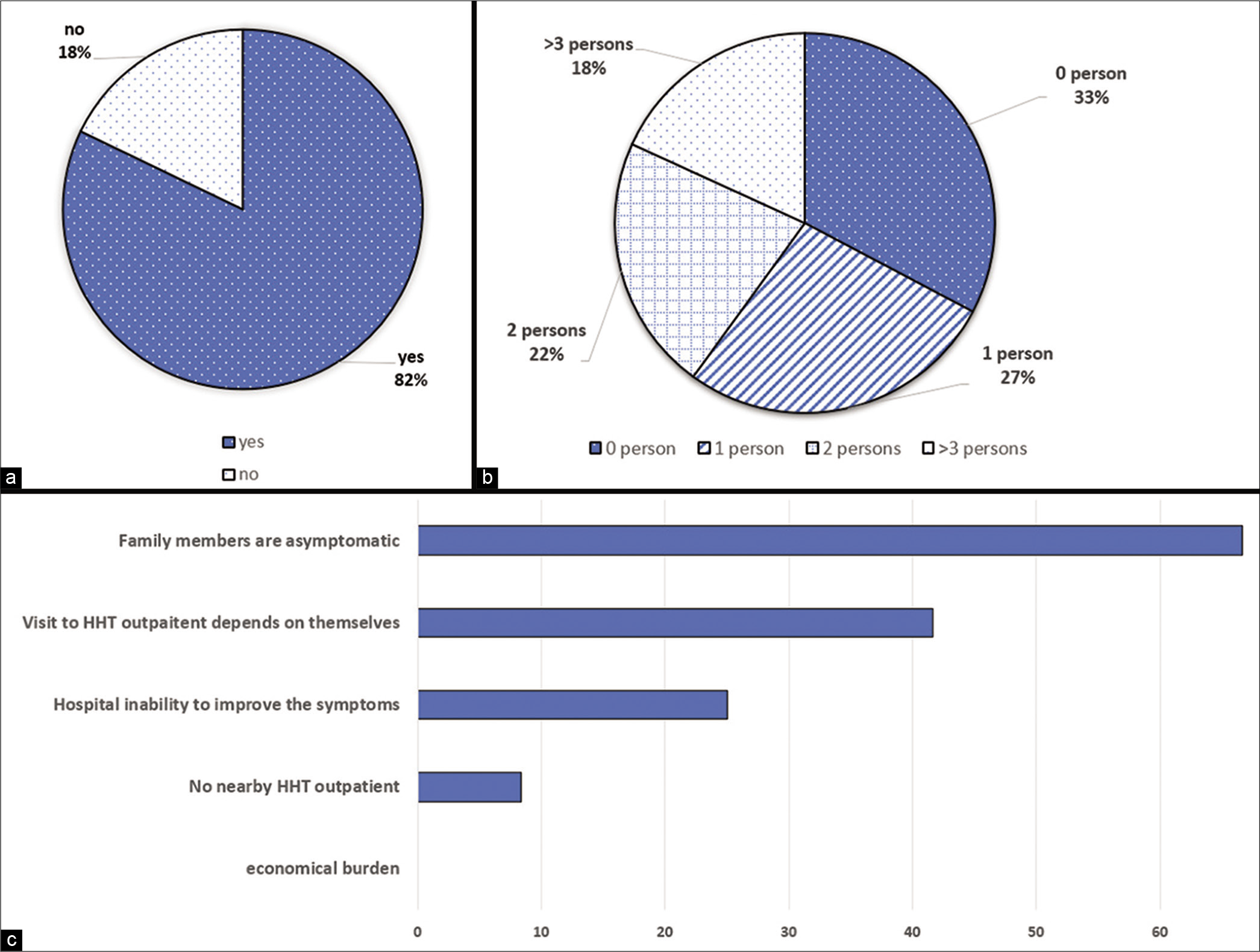
Figure 4:
Data of potential hereditary hemorrhagic telangiectasia (HHT) patients. (a) Proportion of HHT patients who noticed epistaxis in family members. (b) Number of people diagnosed as HHT other than the patient. (c) Reasons why HHT patients do not want their family members not to visit the medical institution.
DISCUSSION
From our survey, current status of HHT medical practice in Japan was elucidated. It was found that to get a confirmed diagnosis of HHT, around 9 years and multiple times screenings in 2–4 hospitals were required as an average. Most patients face economical oppression and have to visit multiple departments to diagnose HHT in addition to the frequent physical anguishing symptoms such as recurrent and intractable nasal bleedings every day. The HHT patients’ reluctance to recommend their family members to visit the doctor could be one of the major reasons why HHT patients have not been diagnosed adequately in Japan.
The road to early diagnosis
Epistaxis is a prominent symptom of HHT.[
How to identify asymptomatic potential HHT patients
HHT is an autosomal dominant disease and the penetrance is almost 100% as the age of patients advanced.[
Future policy for HHT medical management in Japan
HHT shows a wide variety of symptoms because AV shunts are located in many organs such as the liver, intestine, spine, brain, and lungs. Stroke, brain abscesses, hematochezia, hematemesis, and heart failure can sometimes occur and require special treatment by experts.[
There are several potential limitations to this study. The target of this survey is HHT Japan Association’s members and the number of responses was limited. It may be difficult to generalize the study results to the HHT population for the management of the disease in Japan.
CONCLUSION
HHT medical practice in Japan should be further modified and advanced by implementing a social system such as HHT center establishment and education for primary care physicians as well as HHT patients. Such efforts would help alleviate patients’ physical, psychiatric, and economical suffering and could improve the QOL of HHT patients. In addition, appropriate screening administration for potential patients may prolong the overall survival rate of HHT patients in Japan.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
My sincere thanks to Dr. Komiyama who communicated with the members of HHT Japan and helped acquire the necessary data required for this study.
References
1. Berg J, Porteous M, Reinhardt D, Gallione C, Holloway S, Umasunthar T. Hereditary haemorrhagic telangiectasia: A questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003. 40: 585-90
2. Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002. 19: 140-8
3. de Gussem EM, Edwards CP, Hosman AE, Westermann CJ, Snijder RJ, Faughnan ME. Life expectancy of parents with hereditary haemorrhagic telangiectasia. Orphanet J Rare Dis. 2016. 11: 46
4. Dupuis-Girod S, Cottin V, Shovlin CL. The lung in hereditary hemorrhagic telangiectasia. Respiration. 2017. 94: 315-30
5. Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012. 307: 948-55
6. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011. 48: 73-87
7. Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary haemorrhagic telangiectasia (rendu-osler-weber disease). Lancet. 2003. 362: 1490-4
8. Kjeldsen A, Aagaard KS, Tørring PM, Möller S, Green A. 20-year follow-up study of Danish HHT patients-survival and causes of death. Orphanet J Rare Dis. 2015. 11: 157
9. Komiyama M. HHT Japan. Available from: http://www.komiyama.me/HHT_JAPAN/About_US.html [Last accessed on 2020 Jan 12].
10. Li S, Wang SJ, Zhao YQ. Clinical features and treatment of hereditary hemorrhagic telangiectasia. Medicine (Baltimore). 2018. 97: e11687
11. McDonald J, Wooderchak-Donahue W, Webb CV, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front Genet. 2015. 6: 1
12. Morgan T, McDonald J, Anderson C, Ismail M, Miller F, Mao R, Madan A. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (oslerweber-rendu syndrome). Pediatrics. 2002. 109: E12
13. Pierucci P, Lenato GM, Suppressa P, Lastella P, Triggiani V, Valerio R. A long diagnostic delay in patients with hereditary haemorrhagic telangiectasia: A questionnaire-based retrospective study. Orphanet J Rare Dis. 2012. 7: 33
14. Sabbà C, Pasculli G, Suppressa P, D’Ovidio F, Lenato GM, Resta F. Life expectancy in patients with hereditary haemorrhagic telangiectasia. QJM. 2006. 99: 327-34
15. Shovlin CL, Buscarini E, Kjeldsen AD, Mager HJ, Sabba C, Droege F. European reference network for rare vascular diseases (VASCERN) outcome measures for hereditary haemorrhagic telangiectasia (HHT). Orphanet J Rare Dis. 2018. 13: 136
16. Shovlin CL, Condliffe R, Donaldson JW, Kiely DG, Wort SJ. British thoracic society clinical statement on pulmonary arteriovenous malformations. Thorax. 2017. 72: 1154-63
17. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ. Diagnostic criteria for hereditary hemorrhagic telangiectasia (rendu-osler-weber syndrome). Am J Med Genet. 2000. 91: 66-7
18. Shovlin CL. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010. 24: 203-19
19. Tatsumi KInoue GUeda HOsato GKasai TKimura H. Osler Disease (Appointed Number 227) Japan Intractable Diseases Information Center. Available from: https://www.nanbyou.or.jp/entry/4352 [Last accessed on 2020 Feb 04].

