

- Department of Neurosurgery, Ehime University School of Medicine, Toon, Ehime, Japan.
- Department of Pediatrics, Ehime University School of Medicine, Toon, Ehime, Japan.
- Department of Diagnostic Pathology, Ehime University School of Medicine, Toon, Ehime, Japan.
Correspondence Address:
Akihiro Inoue, Department of Neurosurgery, Ehime University School of Medicine, Toon, Ehime, Japan.
DOI:10.25259/SNI_299_2024
Copyright: © 2024 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Masahiro Nishikawa1, Akihiro Inoue1, Kyoko Moritani2, Mari Kagajo2, Riko Kitazawa3, Takeharu Kunieda1. An extremely rare case of primary alveolar rhabdomyosarcoma in the central nervous system. 28-Jun-2024;15:221
How to cite this URL: Masahiro Nishikawa1, Akihiro Inoue1, Kyoko Moritani2, Mari Kagajo2, Riko Kitazawa3, Takeharu Kunieda1. An extremely rare case of primary alveolar rhabdomyosarcoma in the central nervous system. 28-Jun-2024;15:221. Available from: https://surgicalneurologyint.com/surgicalint-articles/12966/
Date of Submission
18-Apr-2024
Date of Acceptance
01-Jun-2024
Date of Web Publication
28-Jun-2024
Abstract
Background: Alveolar rhabdomyosarcoma (ARMS) shows a predilection for the peripheral extremities and is very rarely identified as a primary in the brain. Here, we report a case of ARMS with multiple lesions exclusively within the central nervous system (CNS).
Case Description: A 20-year-old man presented to our hospital with a gradually increasing headache and disturbance of consciousness. Neuroimaging showed hydrocephalus and multiple tumor lesions, including in the brainstem and cerebellum, with uniform gadolinium enhancement on T1-weighted magnetic resonance imaging, as well as spinal cord seeding. Cerebrospinal fluid (CSF) analysis showed a slightly elevated cell count (6/μL; normal,
Conclusion: This report describes an extremely rare case of ARMS arising exclusively within the CNS.
Keywords: Alveolar rhabdomyosarcoma, Central nervous system, H3F3A p.Lys28Met mutation, Myogenin, Spinal dissemination
INTRODUCTION
Rhabdomyosarcoma (RMS) is an aggressive neoplasm characterized by rapid growth and metastatic invasion.[
CASE DESCRIPTION
A 20-year-old man with no relevant medical history presented to our department with a 1-week history of progressive headache and nausea. Intracranial computed tomography revealed marked ventricular enlargement [
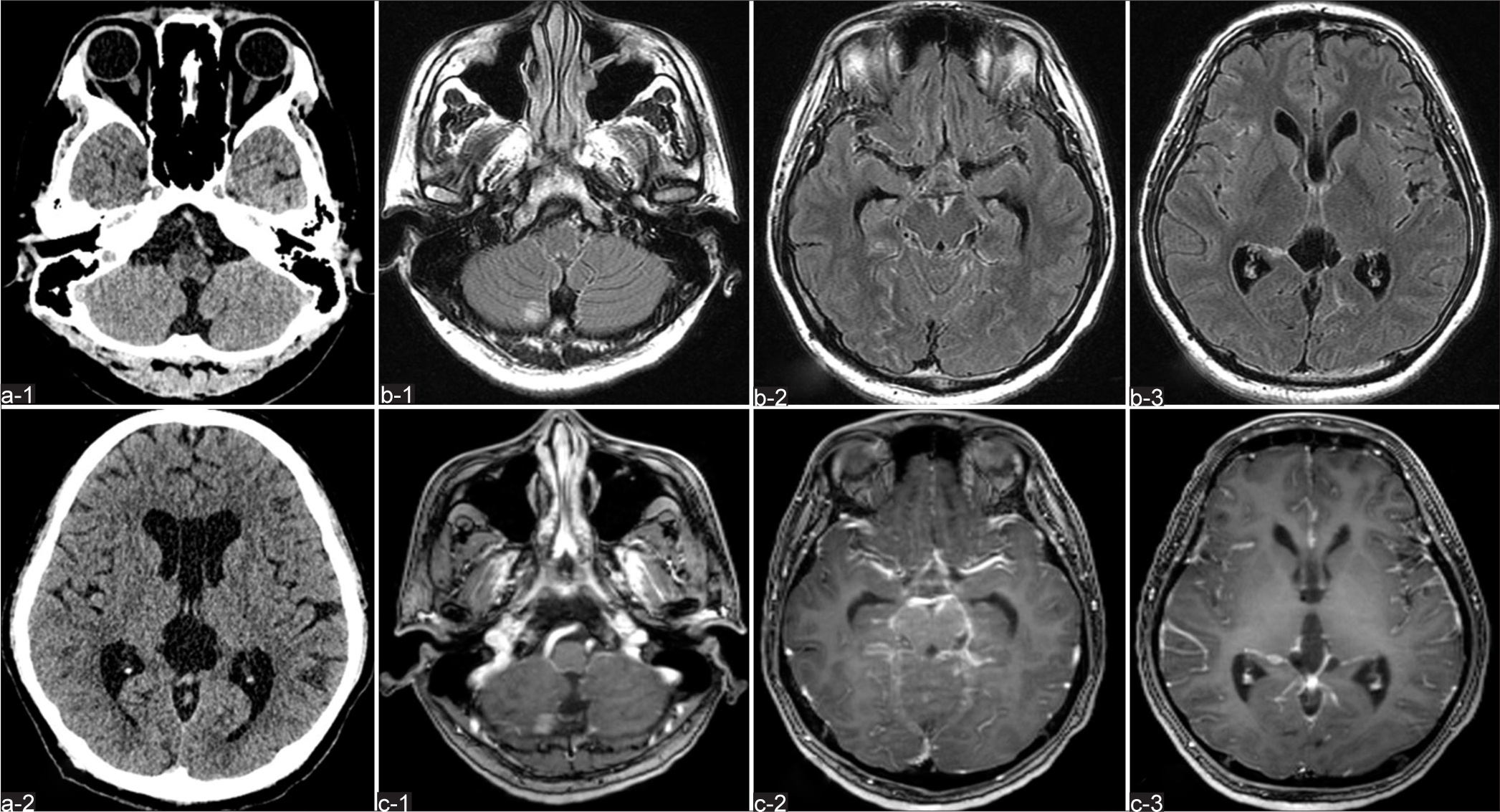
Figure 1:
(a-1, a-2) Computed tomography (CT) without contrast enhancement shows no abnormal masses, but marked ventricular enlargement. Preoperative (b-1, b-2, b-3) axial fluid-attenuated inversion recovery and (c-1, c-2, c-3) gadolinium (Gd)-enhanced T1-weighted imaging (WI) on MRI reveal multiple intracranial areas of abnormal intensity.
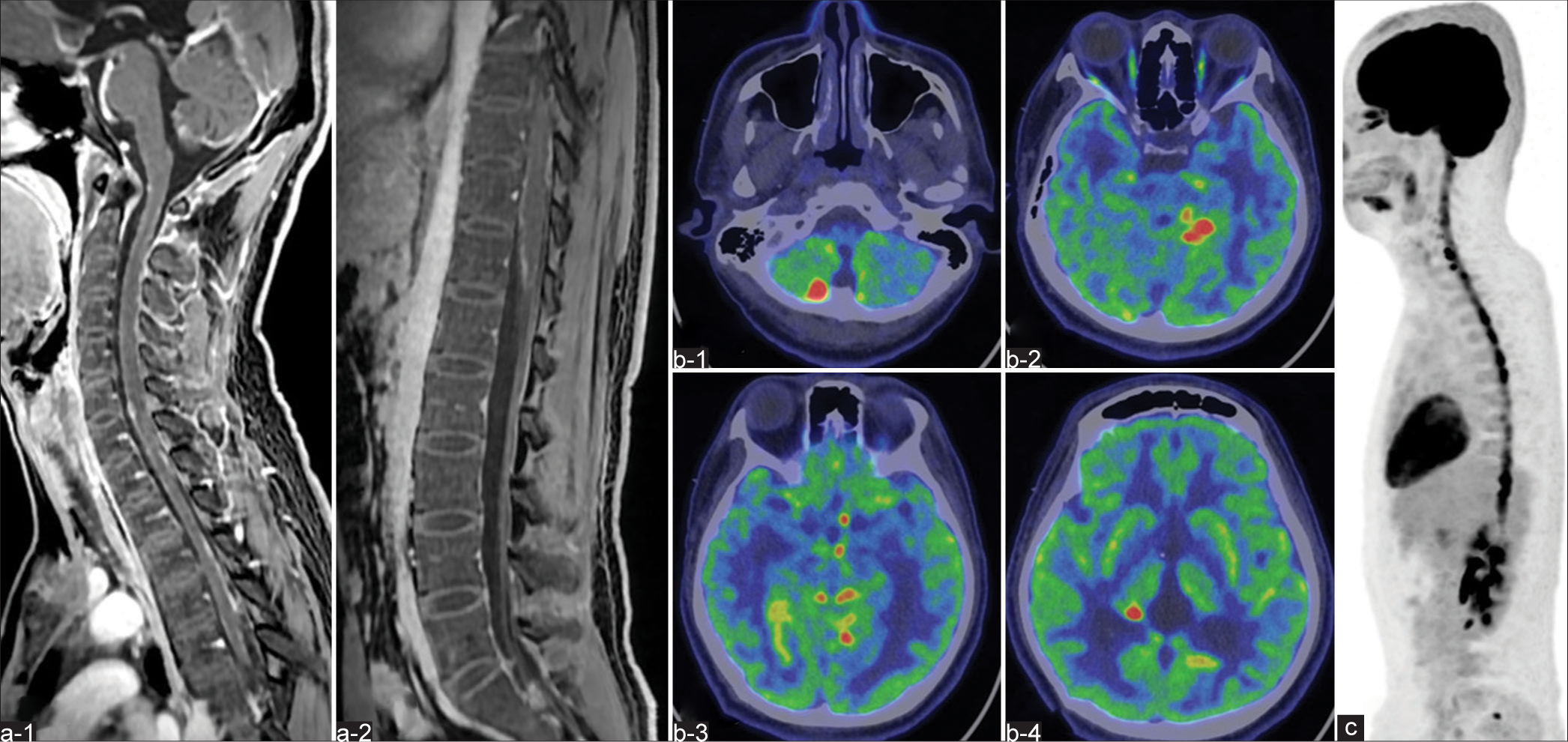
Figure 2:
(a-1, a-2) Magnetic resonance imaging (MRI) demonstrates a longitudinally extending intradural lesion within the spinal cord with Gd enhancement on T1-WI. On 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET), abnormally high uptakes are evident in the (b-1, b-2, b-3, b-4) cranial and (c) spinal region, representing the same sites identified on MRI.
Pathological findings
Postoperative histopathology obtained using hematoxylin and eosin (HE) staining demonstrated a highly cellular area comprising oval and round cells with eosinophilic cytoplasm [
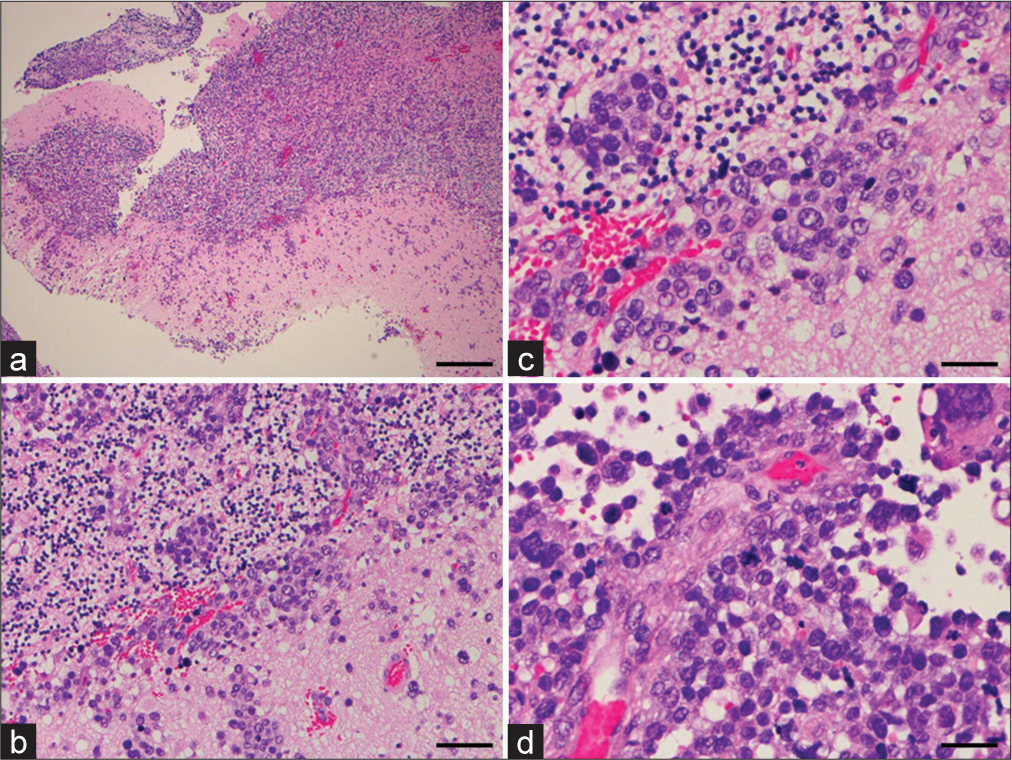
Figure 3:
Histopathology of the biopsy specimen. Hematoxylin and eosin staining show a fascicular arrangement of oval cells with eosinophilic cytoplasm and tumor cells with pleomorphic nuclei and prominent nucleoli. Mitotic figures are elevated, and scattered necrotic foci are apparent. (a) Magnification, ×40; scale bar, 500 μm. (b) Magnification, ×200; scale bar, 100 μm. (c and d) Magnification, ×400; scale bar, 250 μm.

Figure 4:
Photomicrographs revealing the tumor histopathology. (a)Immunohistochemically, tumor cells are negative for the glial marker glial fibrillary acidic protein (GFAP). Negative results are obtained for (b) smooth muscle antigen, but (c) myogenin, and (d) desmin are positive. Immunohistochemical studies also show negative results for (e) S-100 and (f) integrase interactor 1, excluding the diagnoses of malignant peripheral sheath tumor and atypical teratoid/rhabdoid tumor (AT/RT). Magnification, ×400; scale bar, 100 μm.
DISCUSSION
RMS is a common tumor of the head and neck in children and rarely in adults. Among adults, RMS is more common in males (male: female ratio 1.6:1) with a peak age at onset of 44–58 years.[
Whereas previous classifications have divided tumors into embryonal and alveolar subtypes based on histomorphology, the 2020 Classification of Soft-Tissue Tumors by the World Health Organization now divides RMS into embryonal (including the former botryoid category), sclerosing and spindle cell (with myogenic differentiation one mutation or nuclear receptor coactivator two rearrangements), alveolar (with forkhead in RMS rearrangements), and pleomorphic subtypes.[
The majority of case reports have described secondary brain RMS with primaries in the extremities, lungs, scalp, or genital tract. A few cases of primary intracranial ARMS have been reported. A review of the literature identified 48 cases of primary cerebral RMS, among which 33 involved children and the majority of tumor lesions were embedded in the cerebral surface and only rarely entirely intraparenchymal or intraventricular.[
The optimal treatment strategy following histological diagnosis and staging of primary brain ARMS remains contentious. The inherent difficulty in identifying the cell of origin in RMS makes the selection of appropriate chemotherapy difficult and the attendant benefits controversial.[
CONCLUSION
The present case describes one of the few adult cases of primary brain ARMS. Given the rarity of this pathology, a multidisciplinary approach is indispensable. At present, surgical resection of the tumor followed by adjuvant chemotherapy and radiotherapy remains the logical approach to tumor management to maximize long-term survival. While multidisciplinary treatment with chemotherapy and radiotherapy in addition to surgery was performed as early as possible in the present case, good clinical results were not obtained. Further studies and accumulation of cases are therefore needed to understand the behavior of these tumors better, identify an optimal therapeutic plan, and standardize diagnostic immunohistochemical and genetic analyses.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Agamanolis DP, Dasu S, Krill CE. Tumors of skeletal muscle. Hum Pathol. 1986. 17: 778-95
2. Agaram NP. WHO classification of tumours: Soft tissue and bone tumours. Lyon: IARC Press; 2020. p. 211-3
3. Al Mahmasani L, Najjar M, Hourany R, Tabbarah A, Sinno S, Chamseddine N. Primary alveolar rhabdomyosarcoma of the brain: A case report. J Med Case Rep. 2024. 18: 178
4. Ananthaneni A, Kuberappa PH, Srinivas GV, Kiresur MA. Alveolar rhabdomyosarcoma of maxilla. J Oral Maxillofac Pathol. 2016. 20: 164
5. Caporlingua F, Lapadula G, Antonelli M, Missori P. Pleomorphic rhabdomyosarcoma of the cerebellopontine angle in an adult: A review of literature. BMJ Case Rep. 2014. 30: bcr2013203257
6. Celli P, Cervoni L, Maraglino C. Primary rhabdomyosarcoma of the brain: Observations on a case with clinical and radiological evidence of cure. J Neurooncol. 1998. 36: 259-67
7. Ferrari A, Dileo P, Casanova M, Bertulli R, Meazza C, Gandola L. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer. 2003. 98: 571-80
8. Gaiger AM, Soule EH, Newton WA. Pathology of rhabdomyosarcoma: Experience of the intergroup rhabdomyosarcoma study, 1972-78. Natl Cancer Inst Monogr. 1981. 56: 19-27
9. Hollowood K, Fletcher CD. Rhabdomyosarcoma in adults. Semin Diagn Pathol. 1994. 11: 47-57
10. Kelly KM, Womer RB, Sorensen PH, Xiong QB, Barr FG. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol. 1997. 15: 1831-6
11. Khalatbari MR, Hamidi M, Moharamzad Y. Primary alveolar rhabdomyosarcoma of the brain with long-term survival. J Neurooncol. 2013. 115: 131-3
12. Morgenstern D, Rees H, Sebire N, Shipley J, Anderson J. Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: Tissue array study and review of the literature. Pathol Oncol Res. 2008. 14: 233-8
13. Palta M, Riedel RF, Vredenburgh JJ, Cummings TJ, Green S, Chang Z. Primary meningeal rhabdomyosarcoma. Sarcoma. 2011. 2011: 312802
14. Pasquier B, Couderc P, Pasquier D, Hong PM, Pellat J. Primary rhabdomyosarcoma of the central nervous system. Acta Neuropathol. 1975. 33: 333-42
15. Rodriguez-Beato F, De Jesus O, Sanchez-Ortiz J, Delgado P, Perez-Berenguer JL, Labat EJ. Alveolar rhabdomyosarcoma metastatic to the brain. BMJ Case Rep. 2021. 14: e240516
16. Shimoyama R, Sasaki S, Nishikawa K, Matsui M, Fujinaka H, Shimazu K. A case of alveolar rhabdomyosarcoma. J Jpn Soc Clin Cytol. 2015. 54: 41-6
17. Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: A critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011. 145: 967-73


