

- Department of Neurosurgery, Azienda Ospedaliero Universitaria di Sassari, Sassari, Italy.
- Department of Oncology, Azienda Ospedaliero Universitaria di Sassari, Sassari, Italy.
Correspondence Address:
Domenico Policicchio, Department of Neurosurgery, Azienda Ospedaliero Universitaria di Sassari, Sassari, Italy.
DOI:10.25259/SNI_1255_2021
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Domenico Policicchio1, Riccardo Boccaletti1, Angelo Salvatore Cuccu2, Gina Casu1, Giosuè Dipellegrini1, Artan Doda1, Giampiero Muggianu1, Filippo Veneziani Santonio1. Atypical and aggressive diffuse leptomeningeal glioneuronal tumor in a young adult: A case report and review of the literature. 20-May-2022;13:214
How to cite this URL: Domenico Policicchio1, Riccardo Boccaletti1, Angelo Salvatore Cuccu2, Gina Casu1, Giosuè Dipellegrini1, Artan Doda1, Giampiero Muggianu1, Filippo Veneziani Santonio1. Atypical and aggressive diffuse leptomeningeal glioneuronal tumor in a young adult: A case report and review of the literature. 20-May-2022;13:214. Available from: https://surgicalneurologyint.com/surgicalint-articles/11601/
Date of Submission
22-Dec-2021
Date of Acceptance
25-Apr-2022
Date of Web Publication
20-May-2022
Abstract
Background: DLGNT is a rare tumor, commonly diagnosed in pediatric age; in most cases, the pathology presents a slow and indolent evolution. We present a case report of a young adult affected by DLGNT characterized by aggressive and atypical behavior.
Case Description: A 21-year-old male presented with mild paraparesis and hypoesthesia with a D2 level. MRI scan of the brain and spine showed a dorsal intramedullary lesion; a diffuse craniospinal leptomeningeal thickening was also present. After a week, the neurological status deteriorated rapidly with paraparesis worsening and onset of acute hydrocephalus. The patient underwent external ventricular drain positioning; a C7-D4 laminectomy was subsequently performed with partial tumor resection. Histological examination revealed a DLGNT with aggressive aspects (Ki67 30%). Postoperatively, the patient showed an immediate mild worsening of the lower limbs deficit. After a few days, severe further neurological deterioration occurred with progressive motor deficit to the upper limbs and ultimately respiratory failure. Mechanical ventilation was necessary and the patient was transferred to the ICU; during the following weeks, he developed tetraplegia and underwent ventriculoperitoneal shunt positioning. By the time, the histological diagnosis was available, the clinical status would not allow radiotherapy or chemotherapy. The patient deceased approximately 90 days after hospitalization due to respiratory complications.
Conclusion: DLGNT is a rare tumor; diagnosis requires a high index of suspicion and confirmation with biopsy. Although most cases have an indolent course, some patients may have aggressive forms. High proliferation index, hydrocephalus occurrence, and massive craniospinal leptomeningeal spread appear to be associated with worse prognosis.
Keywords: Brain tumor, Central nervous system tumor, Diffuse leptomeningeal glioneuronal tumor, DLGNT, Spine tumor
INTRODUCTION
The diffuse leptomeningeal glioneuronal tumor (DLGNT) is a rare neoplasm. Described as a distinguished entity in the 2016 WHO classification of tumors of the central nervous system (CNS),[
The WHO has not yet assigned a grade to this neoplasm due to the limited number of cases described and clinical, treatment, and outcome heterogenicity. Moreover, the literature data are insufficient to formulate guidelines or suggest a standard management[
A young adult affected by DLGNT characterized by aggressive and atypical behavior was recently treated in our center, with rapidly evolving poor prognosis. We describe the case and report a review of the literature.[
MATERIALS AND METHODS
Case description
In June 2020, a 21-year-old male was admitted to our department after 2 weeks onset of headache, mild paraparesis (right side worse than left side), tactile, and thermic hypoesthesia with a D2 level. A contrast-enhanced MRI scan of the brain and spine showed an intramedullary lesion extending from D1 to D5. The cranial portion of the tumor presented an apparently exophytic extramedullary component which extended on the right posterolateral side up to C7 [
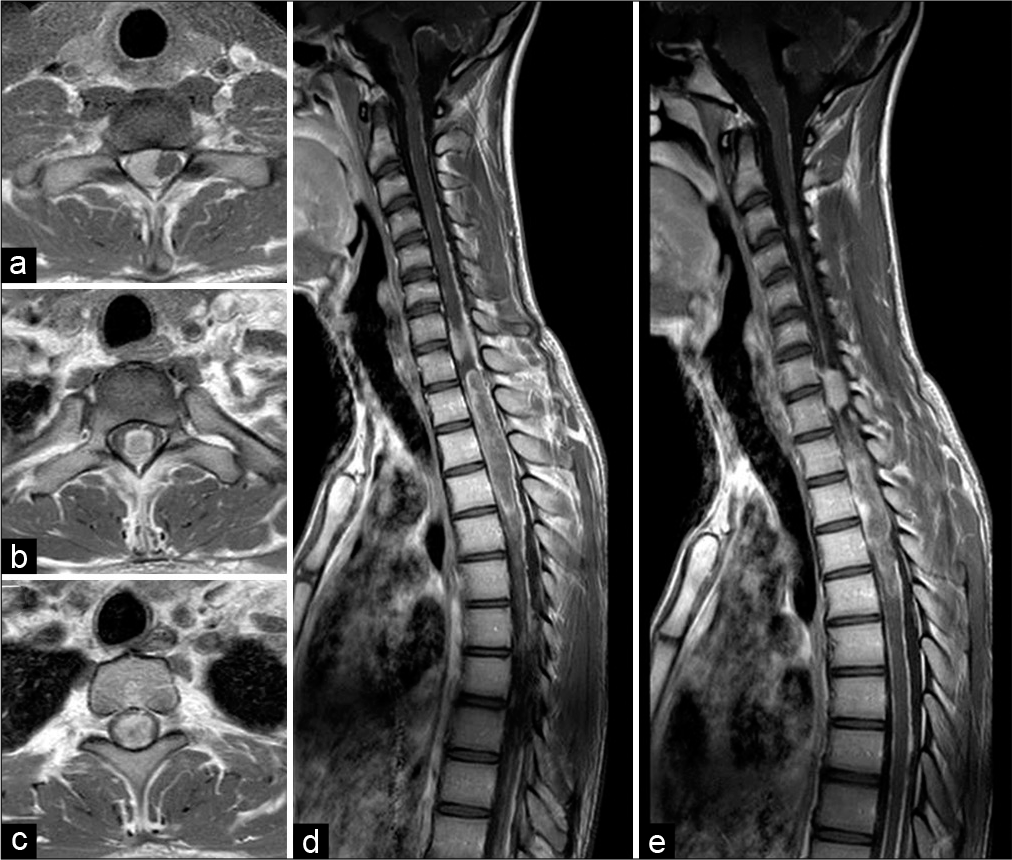
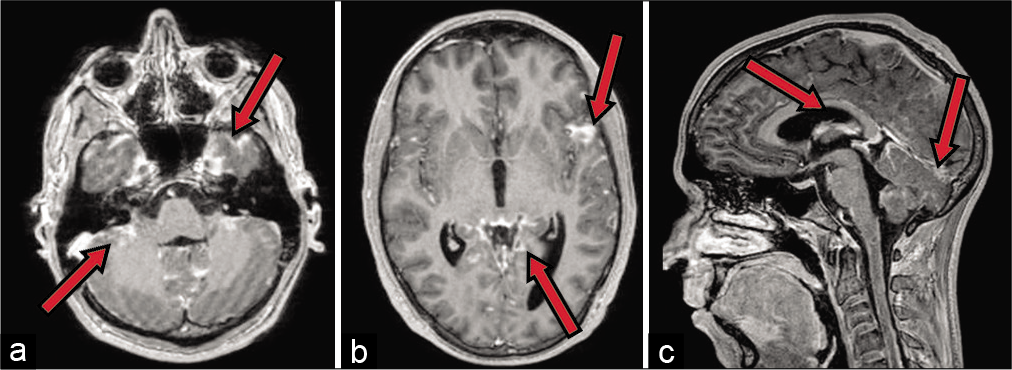
Postoperatively, the patient showed an immediate mild worsening of the lower limbs deficit. After a few days, severe further neurological deterioration occurred with progressive motor deficit to the upper limbs and ultimately respiratory failure. Mechanical ventilation was necessary and the patient was, therefore, transferred to the Intensive Care Unit. During the following weeks, he developed tetraplegia and underwent surgery for ventriculoperitoneal shunt positioning. By the time, the histological diagnosis was available; the clinical status would not allow radiotherapy (RT) or chemotherapy (ChT). The patient deceased approximately 90 days after hospitalization due to respiratory complications.
Histopathological findings
At histological examination, the lesion showed sections of solid neoplasm consisting of small cells proliferation with scarce cytoplasm and round nucleus and larger cells with light cytoplasm, small nucleus, and chromatin granules. The cells were arranged in perivascular aggregates and showed oligodendroglial-like aspects. Immunohistochemistry showed positivity for CD65, GFAP, S100 protein, synaptophysin, olig2 and negativity for EMA, CD45, IDH1 and BRAF V600E mutation, negative BRAF fusion, absence of 1p deletion, or 1p/19q codeletion. Ki-67 was 30%. Diagnosis was consistent with DLGNT.
We conducted a literature review of prior reported case report and case series dealing with DLGNT to identify potential prognostic factors and treatment strategies.
The search was limited to papers in English language published between 2009 and 2020. The only papers considered eligible were those containing a clear description of the clinical, radiological, and pathological features as well as the type of treatment, the outcome, and a minimum 6-month follow-up (FU) (except for cases with lower survival).
Data regarding the patients’ demographic characteristics, symptoms at clinical onset, neuroimaging, presence/absence of hydrocephalus, presence/absence of intraparenchymal lesions, molecular features, type of treatment (surgery, ChT, and RT), outcome, and FU length were collected from the selected papers.
We analyzed the potential prognostic significance of age, radiological appearance (leptomeningeal thickening/ intraparenchymal nodules), hydrocephalus, proliferation index (PI) (Ki67), molecular features (BRAF status, 1p deletion, and 1p/19q codeletion), and type of treatment.
RESULTS
The literature search yielded 26 articles dealing with DLGNT that clearly met our inclusion criteria:[
All data regarding the patients are represented in [
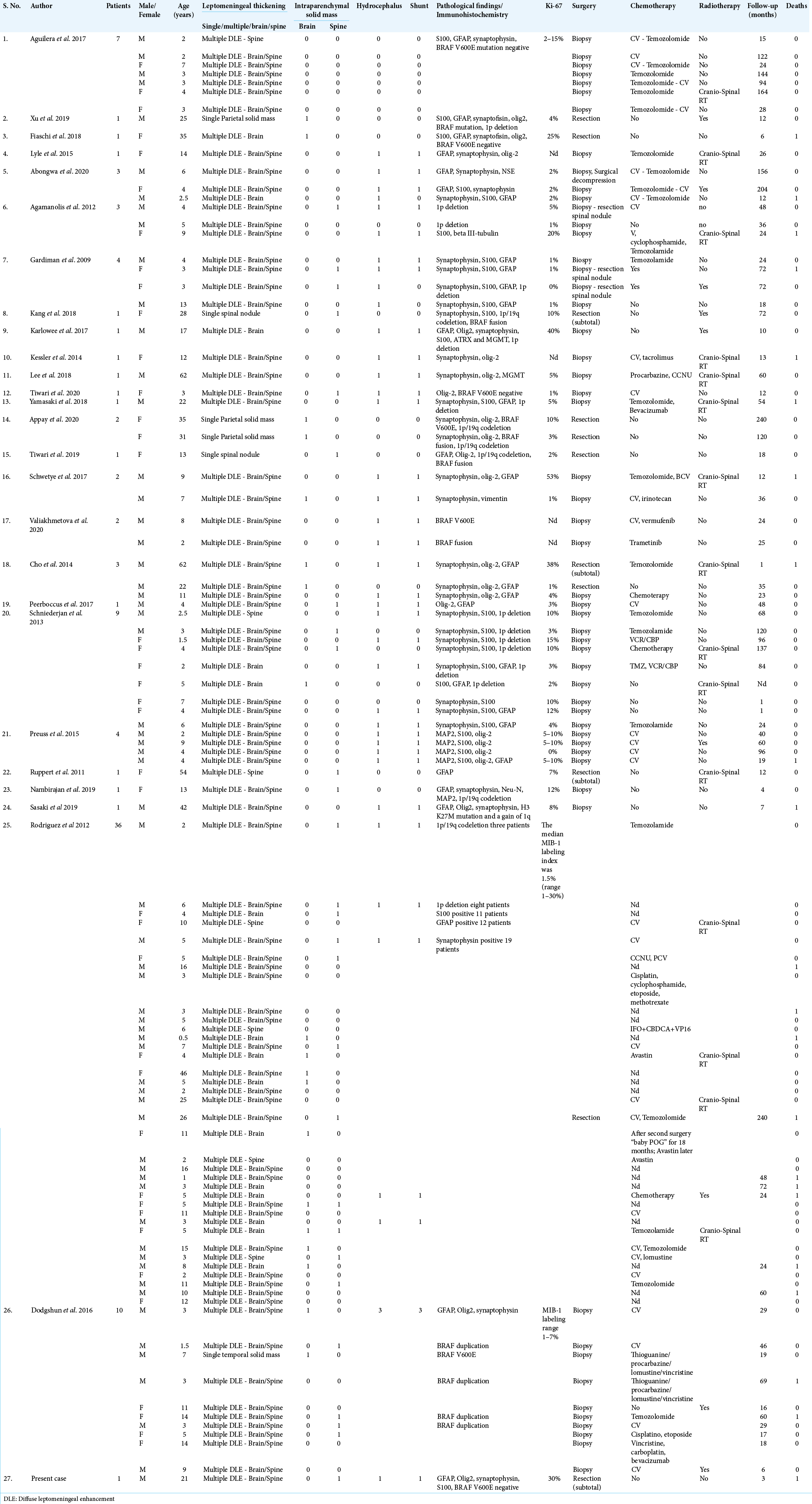

The case histories comprehend 100 patients, 61 males and 39 females.
Average FU of the whole group was 53 months, range 1–240 months. In 22 cases, the patients died because of the disease, average OS 43 months (range 1–240); a patient who die after 20 years was included in the group of patients with better prognosis [
Age at diagnosis
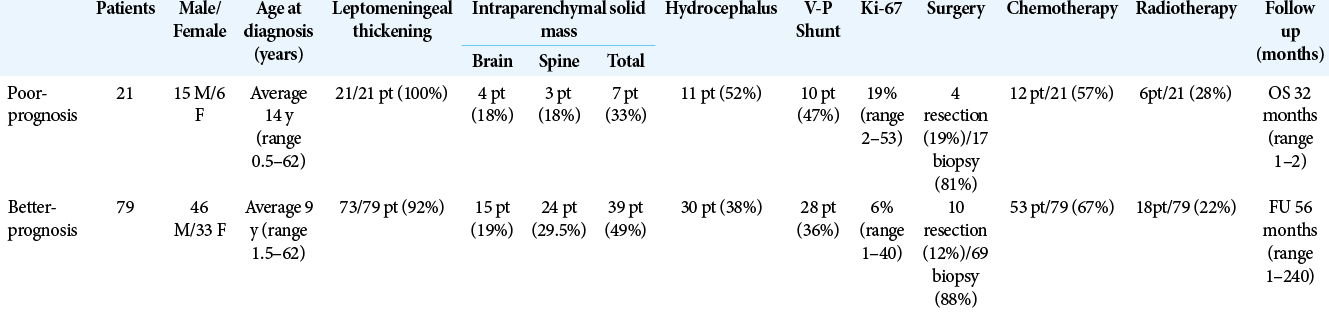
Average age at diagnosis was 10.5 years, median 5 years, and mode 3 years. Minimum age at clinical onset was 0.5 years and maximum age was 62 years. Overall eight patients were older than 30 (adults), seven were between 18 and 30 (young adults), 17 were between 11 and 17 (teenagers), and 68 were between 0 and 10 (children). In the children group, 51 patients were between 0 and 5 and 17 patients were over 5.
Average age at diagnosis resulted higher in the poor-outcome group (14 years vs. 9 years). Mortality 16% (10pt/62) in patients aged 0–8 years versus mortality 32% (12pt/38) in patients older than 8 years.
Clinical onset
Signs and symptoms at clinical onset were heterogeneous and appeared to correlate with the location and extension of the tumor. Signs and symptoms related to hydrocephalus were the most common. Headache is reported as the initial symptom in 45% of cases, both isolated or associated with nausea/vomiting and decreased level of consciousness. Seizures occurred in 15% of cases. Spinal cord or radicular compression syndromes (paraparesis, tetraparesis, ataxia, and radiculopathy) were described in 14% of patients. Meningismus (rigor nucalis, radicular irritation, and photophobia) was present in 10% of cases. Cerebellar syndromes, visual disturbances, aphasia, hemiparesis, scoliosis, pain, behavior disorders, and cranial nerves deficits were also described with the lower incidence.
Based on clinical presentation, no substantial differences were appreciated between the two outcome groups.
Neuroimaging
The typical brain and spine DLE features on MRI were reported in 72 patients. In 15 cases, meningeal involvement was present only at the cerebral level while in seven cases, it was limited to the spine. Altogether 46 solid intraparenchymal lesions were found, 19 brain nodules and 27 spinal nodules. Forty solid lesions were seen in conjunction with the DLE. A single intraparenchymal lesion without DLE was present in six cases (four encephalic and two spinal nodules).
DLE was described in all cases of poor outcome and in 73 of the living patients. Solid intraparenchymal lesions were slightly predominant between the cases with favorable outcome [
Hydrocephalus
Hydrocephalus was observed in 41 patients and a ventriculoperitoneal shunt (VPS) was necessary in 38 cases. Hydrocephalus was more frequent in the poor-outcome group (52% vs. 38%). Mortality rate resulted higher in cases with hydrocephalus (27%, OS 21.9 months range 1–72) respect to cases without hydrocephalus (17%; OS 72 months, range 6–240).
PI
Ki-67 PI ranged from 0% to 53% with an average 9% and a median 4%. In three case series[
Molecular analysis
The molecular features appeared very heterogeneous since not all the papers included in the review report the same type of molecular study. Immunohistochemistry showed positivity for Synaptophysin (36%), Olig-2 (22%), GFAP (29%), and S100 (27%). BRAF V600E mutation in four patients, KIAA1549 BRAF fusion in four patients, BRAF duplication in four patients; BRAF V600E mutations were negative in nine patients and BRAF status was not specified in the remaining 78 patients. 1p deletion was found in 20 cases and 1p/19q codeletion in eight cases while it was not specified in 72 cases. A proper comparison of the molecular analysis (with regard to outcome) was not possible due to the heterogeneity of the molecular profiles in the different studies.
Treatment
Surgical treatment of solid lesions was described in 14 cases. Subtotal (4) or gross total (10) resection of eight spinal and six cerebral nodules was performed. The remaining 86 patients underwent brain or spinal cord biopsy. Mortality 35% (5/14pt) in patients submitted to resective surgery (OS 64.5 months, range 1–240); and mortality 10% (9/86pt) in patients submitted to biopsy (OS 30 months, range 7–69).
ChT was administered to 65 patients. Temozolomide (24), carboplatin (31), and vincristine (31) were the most used medications, although no standard protocol exists [
The 65 patients who have undergone ChT had an average FU at last visit of 59 months, range 1–240 months. Twelve patients (18%) in this group died, average OS 50 months (range 1–240). Average FU in patients not treated with ChT (18) was 36 months (range 1–240 months), 3 of them (16%) deceased, average OS 5 months (range 3–7). It was not possible to identify outcome differences based on the different ChT protocols.
Twenty-four patients underwent RT (15 cerebrospinal RT and nine not specified). In six cases, RT was performed alone while in 18 cases, it was associated with ChT. Among the patients treated with RT (24), average FU was 51 months (range 1–204 months); 6 patients (25%) deceased in this group (average OS 21.3 months, range 1–54). Average FU in patients not treated with RT (18) was 53 months (range 3–240 months); 6 patients (8%) died, average OS 19.3 months (range 3–72). Comparing poor-outcome group with better-outcome group, we observed that surgery and ChT were slightly more frequent in the better-outcome group while RT was slightly prevalent among the poor-outcome group [
DISCUSSION
The DLGNT was introduced in the 2016 WHO classification of tumors of the CNS as “a rare glioneuronal neoplasm characterized by predominant and widespread leptomeningeal growth, an oligodendroglial-like cytology, evidence of neuronal differentiation in a subset of cases, and a high rate of concurrent KIAA1549BRAF gene fusions and either solitary 1p deletion or 1p/19q codeletion in the absence of IDH mutation.”[
Most of these tumors show low-grade histological features and indolent clinical behavior. However, cases presenting anaplasia with increased mitotic activity and aggressive clinical behavior have been described. The clinical picture depends on the location and extension of the neoplasm. Overall signs and symptoms are heterogeneous. DLE is a peculiar aspect on MRI, usually involving the spinal cord and basal cisterns. Non-enhancing multifocal cystic lesions in both the brain and spinal cord are also common. These findings are frequent and may be present in conjunction with intraparenchymal solid lesions. Nevertheless, DLGNT does not always show the typical appearance on neuroimaging.
Diagnosis is often complicated since the pathology is rare and presents heterogeneous clinical patterns.[
At the moment, there are no guide-lines or standard of care available, nor the prognostic factors are known. In most cases, the disease behaves as a low-grade tumor with a long overall survival. However, a definitive grading has not been released by the WHO and numerous cases with aggressive clinical course have been described.
A recent study reported that DLGNTs comprise two methylation classes (MCs), DLGNT MC-1 and MC-2, based on genomic DNA methylation profiles.[
Age and clinical presentation
The literature analysis confirmed predilection for pediatric age as 64% of patients were children and the most affected subgroup was 0–5-years-old. Mean age at diagnosis is higher in the poor-outcome group (14 years vs. 9 years), mortality rate resulted higher in patients older than 8 years (32%) respect to younger patients (16%). These findings suggest that an older age at diagnosis appears to correlate with a worse outcome; however, these data are insufficient to establish age at clinical onset as a certain prognostic factor and need further validation.
Clinical presentation is highly variable, depending on disease extension and location. There is not a pathognomonic set of signs and symptoms, which in most cases are related to hydrocephalus and leptomeningeal involvement. A clinical onset with atypical headache, spinal symptoms, and neuropathy is often observed. Clinical pictures did not show definite differences between the two outcome groups.
Radiological features
The typical radiological picture is characterized by DLE which may be misdiagnosed simulating meningoencephalitis, inflammatory diseases, or other disseminated malignant tumors. Non-invasive investigations (neuroimaging, serology, microbiological, and cytochemical CSF analysis) are insufficient for diagnosis. Cytological CSF testing is rarely diagnostic as well, resulting insufficient for cellular typing.[
Although data are insufficient to provide adequate statistics, qualitative analysis suggests that massive leptomeningeal spread at clinical onset may be associated with a worse prognosis. On the other hand, the presence of intraparenchymal lesions seems associated with better prognosis, particularly if in the absence of DLE. These considerations need further verifications and insights.
Hydrocephalus
Patients in the poor-outcome group presented hydrocephalus more frequently (52% vs. 38%) and consequently, a higher number of VPS was performed in this group [
Altogether diagnosis of hydrocephalus, and consequently the need of VPS positioning, in patients affected by DLGNT is associated with worse prognosis.
PI
Preliminary analysis of the literature showed contrasting results about the prognostic role of the PI. Rodriguez et al. presented the results of the largest series in the literature (36 patients).[
Overall, the literature analysis suggests that a high PI is associated with an unfavorable prognosis.
Histological and molecular features (BRAF status, 1p deletion, and 1p/19q codeletion)
The previous literature reports tried to assign a prognostic role to several histopathologic and molecular aspects. Rodriguez et al. suggested that the presence of glomeruloid microvasculature changes have an unfavorable prognostic role.[
It is, therefore, mandatory to collect further data with standardized molecular diagnostic techniques from larger case series.
Treatment
Assessing a prognostic importance to the different therapeutic options has proved a complex task. The reviewed studies reported highly heterogeneous therapeutic protocols and outcomes. Furthermore, we point out 12 cases which did not undergo any adjuvant treatment following surgery (6) or biopsy (6). These patients had an average FU of 40 months (range 1–240) and 25% mortality, similarly to patients treated with postoperative ChT and RT.
Surgery
In the cases with typical diffuse leptomeningeal involvement, the surgical options are limited to brain or spinal cord biopsy. Surgical resection is indicated in case of solid lesions causing mass effect and focal symptoms. The case we described presented DLE and a spinal nodule causing progressive paraparesis for which surgical resection was performed. Unfortunately, the neoplasm showed infiltrating behavior and malignant biology causing poor outcome.
Surgical resection has been performed on 14 of the 100 patients considered in the present review. In eight cases, both DLE and a solid lesion were present while in six cases, no leptomeningeal involvement occurred. The latter underwent surgical resection followed by ChT in two patients and RT in one. Good outcome was observed in all cases, but it must be taken into consideration that this could be related to less aggressive pathologies rather than treatment efficiency.
Even though we observed higher average mortality in patients submitted to resective surgery (39%) respect to only biopsy (10%), we should point out that the two groups have different number of patients (14 vs. 86) making proper comparison unreliable.
Based on these results, it appears that surgical resection has a therapeutic role when treating mass effect solid lesions. Resective surgery should be, therefore, considered part of a combined therapeutic plan which aims at managing the DLGNT as a disseminated organ pathology rather than a focal lesion. Surgery has, therefore, a positive impact on the prognosis quoad valetudinem but does not seem to influence OS.
ChT
Theoretically, ChT should be a valid therapeutic option, since it has a wide effect on the CNS rather than local action. Based on clinical experience and preliminary literature data, several authors have asserted that ChT is an effective treatment for DLGNT.[
Aguilera et al.[
In the present review, ChT was administered in 67% of cases with better outcome and in 57% of cases with poor outcome.
Mortality was reported in 16% of cases which did not undergo ChT and 18% of cases after ChT. This might as well suggest that patients with slowly progressive diseases were not treated while ChT was administered to patients with more aggressive forms.
Patients treated with ChT had a 59-month mean FU compared to 39-month mean FU in the other cases. The data seem to confirm the effectiveness of ChT in stabilizing the disease and improving length of survival. However, these results are not sufficient to assess the validity of ChT due to the limited number of cases and the treatment heterogeneity which prevent an appropriate statistical analysis. Still, many authors recur to ChT for its diffuse effect on the CNS and suggest that clinical trials are necessary to identify a standard of care.
RT
While ChT is widely accepted by most authors even in the absence of standardized protocols, RT’s role remains uncertain. Reports in the literature present partly contrasting results and considerations. Dodgshun et al.[
Data analysis showed that the mean OS was very similar between radiotreated and not radiotreated patients (51 months vs. 53 months). Radiotreated patients had poor prognosis in 25% of cases versus 8% in untreated patients. It is impossible, however, to distinguish if this difference is related to a negative prognostic impact of RT or if RT was administered more frequently in patients with more aggressive diseases. RT should not be recommended as initial treatment considering the absence of sufficient data, the potential side effects, and the evidence of prolonged disease control in untreated patients. Irradiation could be considered as salvage therapy after further tumor progression.
CONCLUSION
The DLGNT is a rare entity and no guidelines or management standard are available nowadays. Clinical and radiological presentation, such as the outcome, can be highly variable. While most cases show slow progression, aggressive clinical behavior is not rare. However, the grading has not yet been assigned to this pathology in the WHO classification.
Analysis of the literature suggests that high PI, hydrocephalus occurrence, massive leptomeningeal spread, and older age at diagnosis have been more frequently associated with poor outcome. The presence of solid intraparenchymal lesions with absent or limited leptomeningeal involvement at clinical onset can be considered a positive prognostic factor.
Literature data are still not sufficient to assign a certain prognostic or diagnostic role to BRAF status, 1p deletion, and 1p/19q codeletion. Biopsy is mandatory for diagnosis which cannot be obtained with noninvasive examinations.
A combined therapeutic approach is recommended consisting in biopsy followed by chemotherapeutic regimes similarly to other LGG. RT’s role remains uncertain, it could be considered as salvage therapy after tumor progression or as a first-line therapy (associated to ChT) in aggressive forms of DLGNT. Surgical resection is reserved in case of solid nodule causing mass effect and neurological impairment and it should not be considered a curative treatment (symptomatic effect). VPS positioning at any stage of the disease is mandatory in case of hydrocephalus, despite a high risk of shunt malfunction.
The observed results need further validation; larger clinical trials conducted with a reliable methodology seem advisable.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abongwa C, Cotter J, Tamrazi B, Dhall G, Davidson T, Margol A. Primary diffuse leptomeningeal glioneuronal tumors of the central nervous system: Report of three cases and review of literature. Pediatr Hematol Oncol. 2020. 37: 248-58
2. Agamanolis DP, Katsetos CD, Klonk CJ, Bartkowski HM, Ganapathy S, Staugaitis SM. An unusual form of superficially disseminated glioma in children: report of 3 cases. J Child Neurol. 2012. 27: 727-33
3. Aguilera D, Castellino RC, Janss A, Schniederjan M, McNall R, MacDonald T. Clinical responses of patients with diffuse leptomeningeal glioneuronal tumors to chemotherapy. Childs Nerv Syst. 2018. 34: 329-34
4. Appay R, Pages M, Colin C, Jones DTW, Varlet P, FigarellaBranger D. Diffuse leptomeningeal glioneuronal tumor: A double misnomer? A report of two cases. Acta Neuropathol Commun. 2020. 8: 95
5. Cho HJ, Myung JK, Kim H, Park CK, Kim SK, Chung CK. Primary diffuse leptomeningeal glioneuronal tumors. Brain Tumor Pathol. 2015. 32: 49-55
6. Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018. 136: 239-53
7. Dodgshun AJ, SantaCruz N, Hwang J, Ramkissoon SH, Malkin H, Bergthold G. Disseminated glioneuronal tumors occurring in childhood: Treatment outcomes and BRAF alterations including V600E mutation. J Neurooncol. 2016. 128: 293-302
8. Fiaschi P, Badaloni F, Cagetti B, Bruzzone L, Marucci G, Dellachà A. Disseminated oligodendroglial-like leptomeningeal tumor in the adult: Case report and review of the literature. World Neurosurg. 2018. 114: 53-7
9. Gardiman MP, Fassan M, Orvieto E, D’Avella D, Denaro L, Calderone M. Diffuse leptomeningeal glioneuronal tumors: A new entity?. Brain Pathol. 2010. 20: 361-6
10. Garibotto F, Pavanello M, Milanaccio C, Gaggero G, Fiaschi P. Management of hydrocephalus related to diffuse leptomeningeal glioneuronal tumour: A multifaceted condition. Childs Nerv Syst. 2021. 37: 1039-40
11. Kang JH, Buckley AF, Nagpal S, Fischbein N, Peters KB. A diffuse leptomeningeal glioneuronal tumor without diffuse leptomeningeal involvement: Detailed molecular and clinical characterization. J Neuropathol Exp Neurol. 2018. 77: 751-6
12. Karlowee V, Kolakshyapati M, Amatya VJ, Takayasu T, Nosaka R, Sugiyama K. Diffuse leptomeningeal glioneuronal tumor (DLGNT) mimicking Whipple’s disease: A case report and literature review. Childs Nerv Syst. 2017. 33: 1411-4
13. Kessler BA, Bookhout C, Jaikumar S, Hipps J, Lee YZ. Disseminated oligodendroglial-like leptomeningeal tumor with anaplastic progression and presumed extraneural disease: Case report. Clin Imaging. 2015. 39: 300-4
14. Lee JK, Ko HC, Choi JG, Lee YS, Son BC. A case of diffuse leptomeningeal glioneuronal tumor misdiagnosed as chronic tuberculous meningitis without brain biopsy. Case Rep Neurol Med. 2018. 2018: 1391943
15. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JP. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ. 2009. 339: b2700
16. Louis DN, Ohgaki H, Wiestier OD, Cavenee WK, Ellison DW, Figarella-Branger D. World Health Organization classification of tumours WHO classification of tumours of the central nervous system. Acta Neuropathol. 2016. 131: 254-7
17. Louis DN, Perry A, Reifenberger G, von Deimling A, FigarellaBranger D, Cavenee WK. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
18. Lyle MR, Dolia JN, Fratkin J, Nichols TA, Herrington BL. Newly identified characteristics and suggestions for diagnosis and treatment of diffuse leptomeningeal glioneuronal/ neuroepithelial tumors: A case report and review of the literature. Child Neurol Open. 2015. 2: 2329048X14567531
19. Nambirajan A, Suri V, Kedia S, Goyal K, Malgulwar PB, Khanna G. Paediatric diffuse leptomeningeal tumor with glial and neuronal differentiation harbouring chromosome 1p/19q co-deletion and H3.3 K27M mutation: Unusual molecular profile and its therapeutic implications. Brain Tumor Pathol. 2018. 35: 186-91
20. Peerboccus M, Beltran-Marin M, Sariban E, Fontanges Q, Ziereisen F. Disseminated oligodendroglial-like leptomeningeal tumor of childhood: A distinctive entity revised and correlated with pathology. J Belg Soc Radiol. 2017. 101: 19
21. Policicchio D, Boccaletti R, Dipellegrini G, Doda A, Stangoni A, Veneziani SF. Pedicled multifidus muscle flap to treat inaccessible dural tear in spine surgery: Technical note and preliminary experience. World Neurosurg. 2021. 145: 267-77
22. Policicchio D, Doda A, Sgaramella E, Ticca S, Veneziani Santonio F, Boccaletti R. Ultrasound-guided brain surgery: Echographic visibility of different pathologies and surgical applications in neurosurgical routine. Acta Neurochir (Wien). 2018. 160: 1175-85
23. Policicchio D, Ticca S, Dipellegrini G, Doda A, Muggianu G, Boccaletti R. Multimodal surgical management of cerebral lesions in motor-eloquent areas combining intraoperative 3D ultrasound with neurophysiological mapping. J Neurol Surg A Cent Eur Neurosurg. 2021. 82: 344-56
24. Preuss M, Christiansen H, Merkenschlager A, Hirsch FW, Kiess W, Müller W. Disseminated oligodendroglial cell-like leptomeningeal tumors: Preliminary diagnostic and therapeutic results for a novel tumor entity. J Neurooncol. 2015. 124: 65-74
25. Reifenberger G, Rodriguez F, Burger PC, Perry A, Capper D, Louis DN, Ohgaki H, Wiestler OD, Cavenee WK.editors. Diffuse leptomeningeal glioneuronal tumor. World Health Organization Histological Classification of Tumours of the Central Nervous System. Lyon: International Agency for Research on Cancer; p. 152-155
26. Richetta C, Shofty B, Gurevich A, Grossman R. Valve failure in ventriculoperitoneal shunts of neuro-oncologic patients: A historical cohort study. World Neurosurg. 2019. 128: e329-33
27. Rodriguez FJ, Perry A, Rosenblum MK, Krawitz S, Cohen KJ, Lin D. Disseminated oligodendroglial-like leptomeningeal tumor of childhood: A distinctive clinicopathologic entity. Acta Neuropathol. 2012. 124: 627-41
28. Ruppert B, Welsh CT, Hannah J, Giglio P, Rumboldt Z, Johnson I. Glioneuronal tumor with neuropil-like islands of the spinal cord with diffuse leptomeningeal neuraxis dissemination. J Neurooncol. 2011. 104: 529-33
29. Sáez-Alegre M, Gutiérrez JM, Contreras CU, Santos FJ, GarcíaFeijoo P, Benito FC. Diffuse leptomeningeal glioneuronal tumour: Where to biopsy? Case report and literature review. Childs Nerv Syst. 2021. 37: 2405-8
30. Sasaki S, Hirose T, Nobusawa S, Myojin T, Morita K, Nakai T. Anaplastic diffuse leptomeningeal glioneuronal tumor associated with H3 K27M mutation. Hum Pathol Case Rep. 2019. 17: 1-4
31. Schniederjan MJ, Alghamdi S, Castellano-Sanchez A, Mazewski C, Brahma B, Brat DJ. Diffuse leptomeningeal neuroepithelial tumor: 9 pediatric cases with chromosome 1p/19q deletion status and IDH1 (R132H) immunohistochemistry. Am J Surg Pathol. 2013. 37: 763-71
32. Schwetye KE, Kansagra AP, McEachern J, Schmidt RE, Gauvain K, Dahiya S. Unusual high-grade features in pediatric diffuse leptomeningeal glioneuronal tumor: Comparison with a typical low-grade example. Hum Pathol. 2017. 70: 105-12
33. Tiwari N, Tamrazi B, Robison N, Krieger M, Ji J, Tian D. Unusual radiological and histological presentation of a diffuse leptomeningeal glioneuronal tumor (DLGNT) in a 13-year-old girl. Childs Nerv Syst. 2019. 35: 1609-14
34. Tiwari S, Yadav T, Pamnani J, Mathew JM, Elhence P, Praneeth K. Diffuse leptomeningeal glioneuronal tumor: A unique leptomeningeal tumor entity. World Neurosurg. 2020. 135: 297-300
35. Valiakhmetova A, Papusha L, Druy A, Yasko L, Ektova A, Karachunsky A. Pediatric diffuse leptomeningeal glioneuronal tumor: Two clinical cases of successful targeted therapy. Pediatr Blood Cancer. 2020. 67: e28478
36. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-Jonathan-Moyal E. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017. 18: e315-29
37. Xu H, Chen F, Zhu H, Luo L, Zhang R. Diffuse leptomeningeal glioneuronal tumor in a Chinese adult: A novel case report and review of literature. Acta Neurol Belg. 2020. 120: 247-56
38. Yamasaki T, Sakai N, Shinmura K, Kawaji H, Koizumi S, Samashima T. Anaplastic changes of diffuse leptomeningeal glioneuronal tumor with polar spongioblastoma pattern. Brain Tumor Pathol. 2018. 35: 209-16

