

- Department of Neurological Surgery, Columbia University Medical Center, New York, NY, USA
- Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA
Correspondence Address:
M. Praver
Department of Neurological Surgery, Columbia University Medical Center, New York, NY, USA
DOI:10.4103/2152-7806.161790
Copyright: © 2015 Praver M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.How to cite this article: Praver M, R. D’Amico, Arraez C, Zacharia BE, Varma H, Goldman JE, Bruce JN, Canoll P. Atypical pleomorphic neoplasms of the pineal gland: Case report and review of the literature. Surg Neurol Int 30-Jul-2015;6:129
How to cite this URL: Praver M, R. D’Amico, Arraez C, Zacharia BE, Varma H, Goldman JE, Bruce JN, Canoll P. Atypical pleomorphic neoplasms of the pineal gland: Case report and review of the literature. Surg Neurol Int 30-Jul-2015;6:129. Available from: http://surgicalneurologyint.com/surgicalint_articles/atypical-pleomorphic-neoplasms-of-the-pineal-gland-case-report-and-review-of-the-literature/
Abstract
Background:Pineal region tumors are rare and diverse. Among them exist reports of pleomorphic xanthroastrocytoma (PXA) and pleomorphic granular cell astrocytoma (PGCA) of the pineal gland. These related tumors are remarkably similar sharing pleomorphic histologic features with only minor immunohistochemical and ultrastructural differences.
Case Description:We present a case of a 42-year old right-handed woman presented with a longstanding history of migraine headaches which had worsened over the two months leading up to her hospitalization. MRI revealed a 1.7 x 1.3 x 1.6 cm intensely enhancing lesion originating in the pineal gland. The tumor closely resembled PGCA but did not strictly fit the diagnostic requirements of either PGCA or PXA.
Conclusion:The present case highlights the exotic nature of pineal region tumors with pleomorphic cell histology. Given the diverse range of tumors encountered in the pineal region, pathological confirmation is mandatory. Favorable clinical outcomes demonstrate that surgical resection alone can yield excellent long-term results for tumors falling within the spectrum of pleomorphic lesions of the pineal gland.
Keywords: Pineal gland, pleomorphic granular cell astrocytoma, pleomorphic xanthoastrocytoma
INTRODUCTION
Pineal region tumors are rare, representing less than 0.5–2% of all intracranial tumors.[
CASE PRESENTATION
A 42-year-old right-handed female presented with a longstanding history of migraine headaches, which had worsened over the 2 months leading up to her hospitalization. She was otherwise healthy with no additional past medical history, taking only rizatriptan and fenoprofen. Her neurological examination was normal.
Magnetic resonance imaging (MRI) revealed a 1.7 × 1.3 × 1.6 cm intensely enhancing lesion originating in the pineal gland and inseparable from the superior aspect of the tectum inferiorly. The lesion appeared slightly hyperintense relative to cerebrospinal fluid (CSF) on T1-weighted images (WI) [
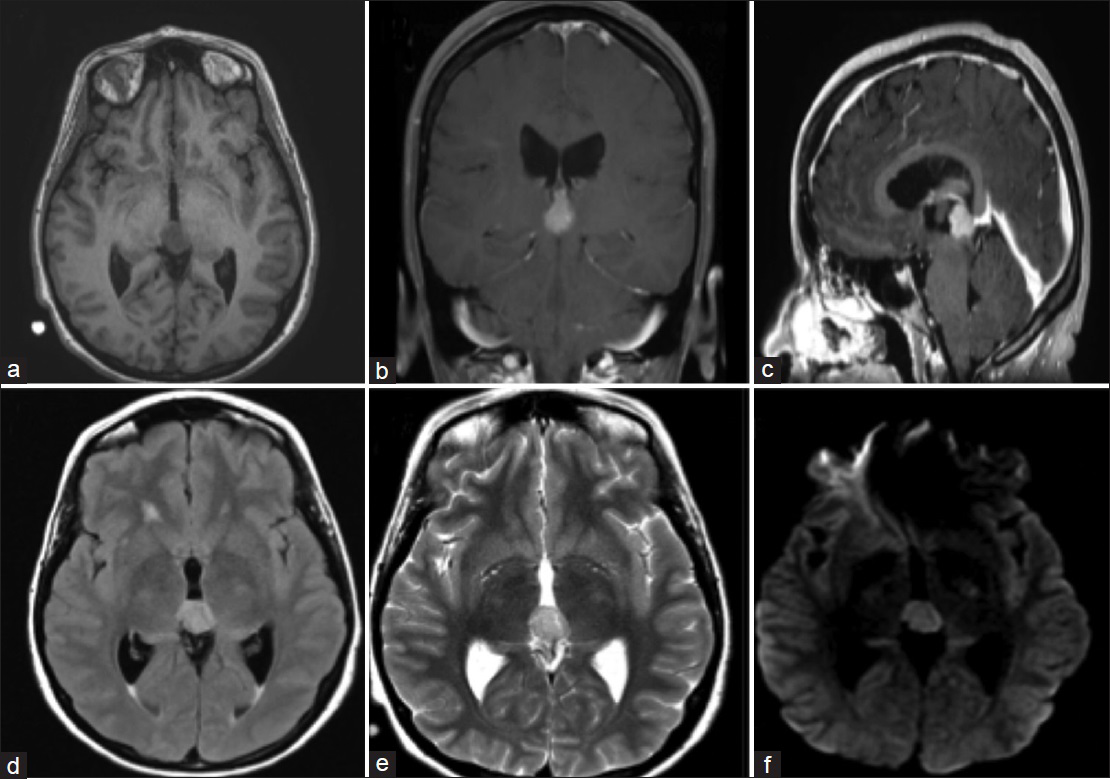
Figure 1
Preoperative Magnetic Resonance Images. (a) Axial T1-weighted image demonstrating lesion in the pineal region with evidence of early hydrocephalus. (b) Coronal contrast enhanced T1-weighted image showing an enhancing lesion extending inferiorly. (c) Sagittal contrast enhanced T1-weighted image demonstrating enhancing lesion displacing the quadrigeminal cistern and dorsal midbrain. (d) Axial FLAIR demonstrating moderate hyperintense lesion. (e) Axial T2-weighted image showing moderate hyperintense lesion. (f) Axial DWI showing moderate hyperintense lesion
The patient initially underwent successful endoscopic third ventriculostomy for relief of obstructive hydrocephalus. CSF glucose and protein were 38 and <10 mg/dL, respectively. Surgical resection took place 18 days later through a supracerebellar, infratentorial approach with the patient in the sitting position.[
Postoperatively the patient did well with a transient mild Parinaud syndrome and gait disturbance that resolved over the following 2 weeks. Follow-up MRI on postoperative day 3 demonstrated complete resection of the tumor [
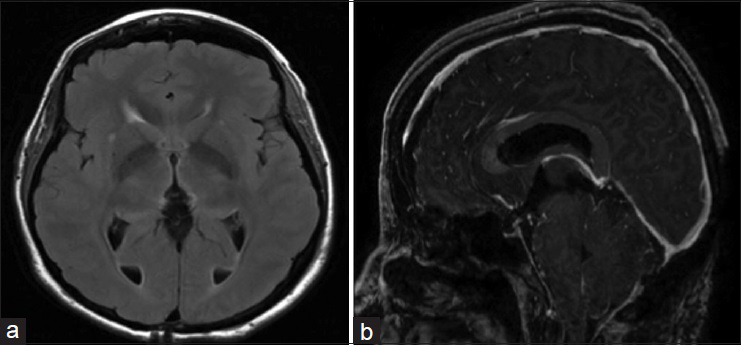
On hematoxylin and eosin stain [
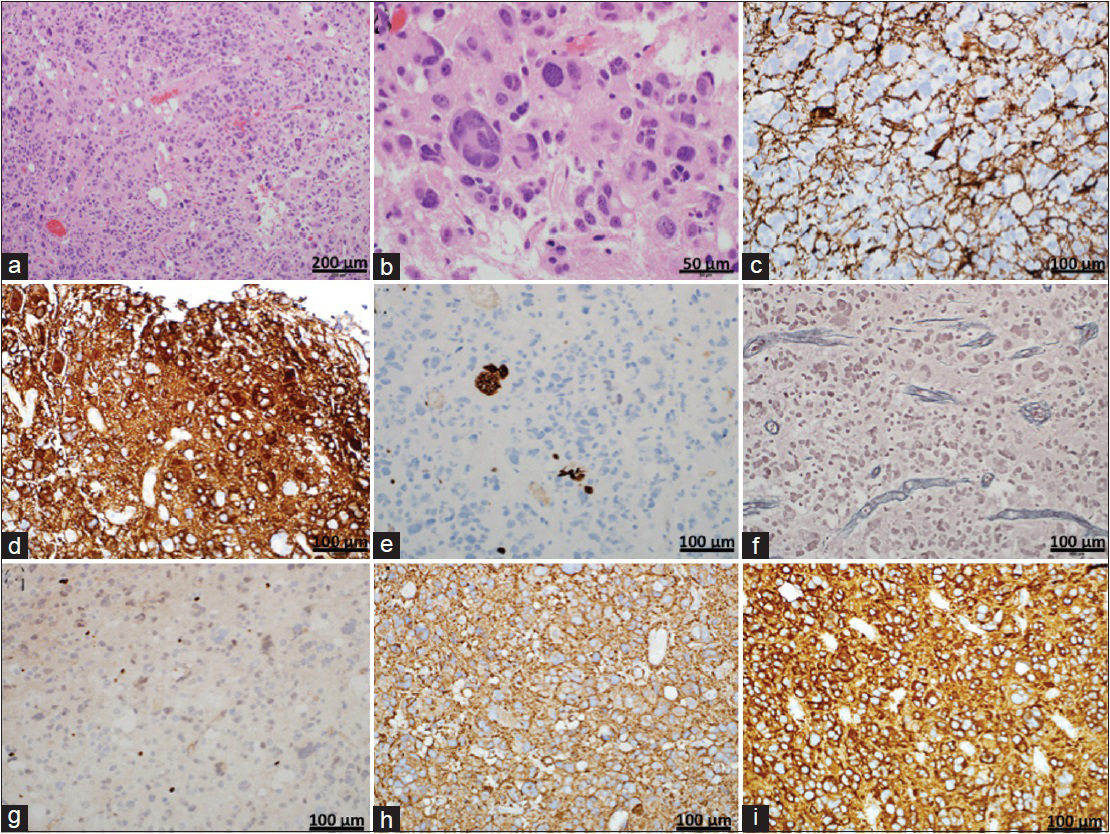
Figure 3
Histology (a and b) Hematoxylin and eosin stained section demonstrating a moderately cellular neoplasm with highly pleomorphic, hyperchromatic nuclei. Frequent multinucleated and enlarged cells with giant, bizarre-shaped nuclei are present. Vessel walls are hyalinized and no areas of vascular proliferation or necrosis are noted. Rare mitotic figures are seen. (c and d) Photomicrograph of GFAP immunostaining demonstrating primarily focal positivity resembling a reactive process with discrete areas of diffuse positive staining. (e) Photomicrograph showing representative Ki-67 immunostaining. Cells show a low proliferation index with focal areas up to 1.6%. (f) Photomicrograph showing representative reticulin immunostaining. Staining can be seen in perivascular connective tissue but there is no reticulin network between the tumor cells. (g) Photomicrograph of Olig2 immunostaining demonstrating scattered positive cells. Magnification: ×20 (h) Photomicrograph of synaptophysin immunostaining demonstrating a diffuse, strongly positive pattern. (i) Photomicrograph of Class III β-tubulin immunostaining demonstrating a diffuse, strongly positive pattern
DISCUSSION
Pineal region tumors are rare, representing 0.5–2% of all intracranial tumors.[
Symptoms of pineal PXA/PGCA at presentation are commonly due to hydrocephalus from mass effect from aqueduct compression and include headache, nausea, and vomiting [
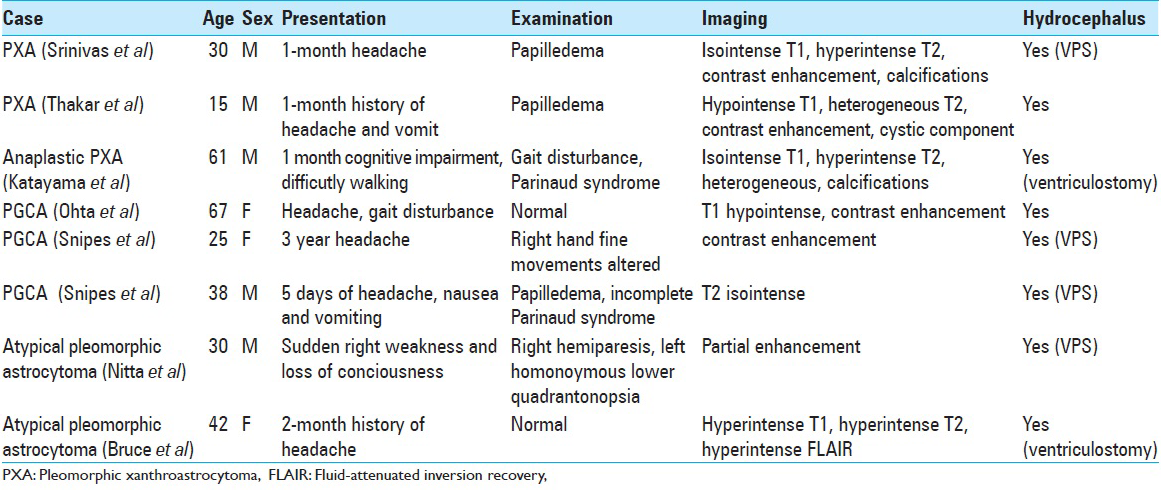
MRI characteristics of pineal PXA/PGCA are variable. Srinivas et al. described an enhancing homogenous lesion with a speck of calcification.[
Histologically, PXA tumor cells appear markedly pleomorphic with bizarre, multinucleated giant cells that vary in size and shape. Intracytoplasmic lipid droplets are often present. There is variable infiltration of the underlying brain parenchyma.[
Pleomorphic granular cell astrocytoma (PGCA) is a tumor with many histologic features that resemble PXA. However, unlike PXA, it has large numbers of mitochondria and does not have reticulin fibers or a basement membrane between adjacent cells.[
Nitta et al. described a pineal gland tumor that could not strictly be defined as PXA or PGCA and was labeled an atypical pleomorphic astrocytoma.[
Similarly, the tumor reported here lacks specific criteria to meet a strict diagnostic category. While it demonstrates pleomorphic nuclei, a strong reticulin network as seen in PXA is lacking. Of even greater peculiarity is the GFAP staining, which, in most areas, resembles a reactive astrocytic process. However, there are discrete areas in which the tumor cells themselves are GFAP positive. This is similar to what was encountered in the atypical pleomorphic astrocytoma described by Nitta et al. While the tumor reported here has features of PXA and of PGCA, perhaps it is best described as a pleomorphic neuroepithelial neoplasm of the pineal gland.
Astrocytes of the pineal gland are largely believed to give rise to these pleomorphic tumors.[
Interestingly, the pathological and immunohistochemical variability seen among PXA, PGCA, and the atypical tumor described here and encountered by Nitta et al. are of less clinical consequence than the overall proliferation indexes of these tumors. All seven reported pleomorphic tumors of the pineal gland shared favorable outcomes [
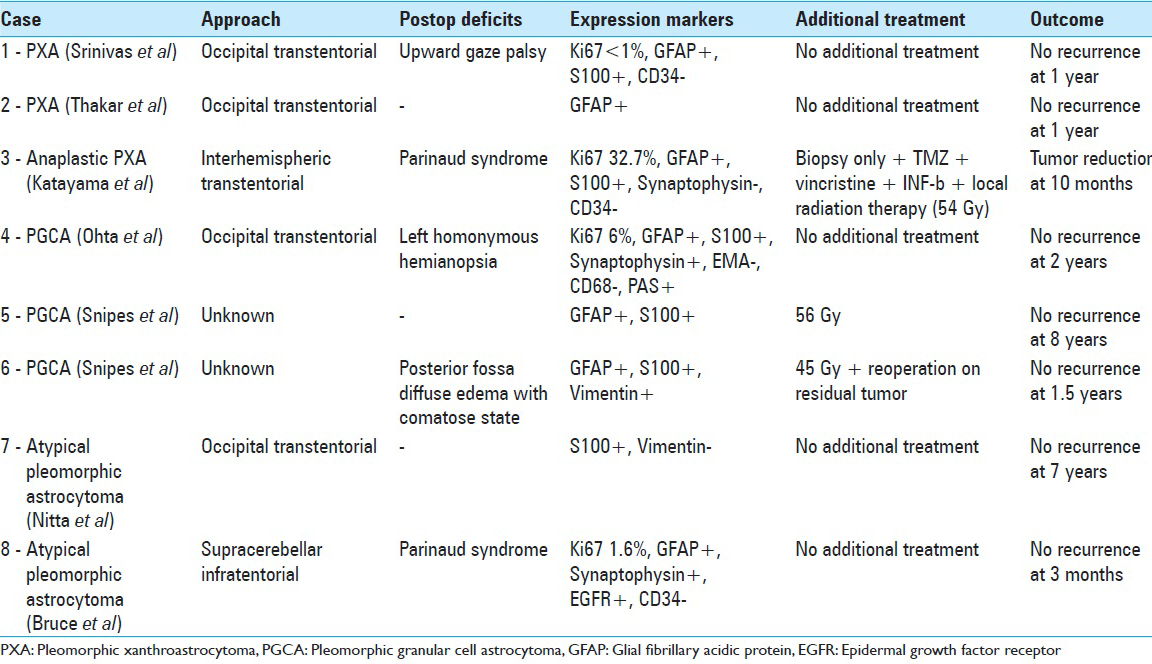
Pineal region tumors span a highly diverse spectrum of histologies ranging from benign to malignant. Accurate histologic diagnosis is essential for optimal clinical management but can be difficult to achieve because of the propensity for mixed tumor pathologies and heterogeneity in the pineal region. In this case, the patient was managed with craniotomy and open microdissection to achieve the goals of definitive diagnosis by maximizing the amount of tissue provided to the pathologists. Open microsurgical procedures have the added benefit of facilitating gross total resection while minimizing the potential for tumor-associated hemorrhage. Alternate approaches including stereotactic biopsy or endoscopic biopsies are acceptable but provide only limited tissue sampling and ignore the benefits of tumor debulking achievable with open resection, especially for benign, encapsulated tumors. While multiple studies have demonstrated safety and efficacy of endoscopic techniques[
CONCLUSION
The present case highlights the exotic nature of pineal region tumors with pleomorphic cell histology. Given the diverse range of tumors encountered in the pineal region, pathological confirmation is mandatory. Favorable clinical outcomes demonstrate that surgical resection alone can yield excellent long-term results for tumors falling within the spectrum of pleomorphic lesions of the pineal gland.
References
1. Abe T, Inoue R, Isono M, Ishii K, Fujiki M, Kamida T. Benign pleomorphic astrocytoma in the hypothalamus-case report. Neurol Med Chir (Tokyo). 2006. 46: 101-3
2. Al-Tamimi YZ, Bhargava D, Surash S, Ramirez RE, Novegno F, Crimmins DW. Endoscopic biopsy during third ventriculostomy in paediatric pineal region tumours. Childs Nerv Syst. 2008. 24: 1323-6
3. Arita K, Kurisu K, Tominaga A, Sugiyama K, Sumida M, Hirose T. Intrasellar pleomorphic xanthoastrocytoma: Case report. Neurosurgery. 2002. 51: 1079-82
4. Blakeley JO, Grossman SA. Management of pineal region tumors. Curr Treat Options Oncol. 2006. 7: 505-16
5. Bruce JN, Stein BM. Surgical management of pineal region tumors. Acta Neurochir. 1995. 134: 130-5
6. Bucciero A, De Caro M, De Stefano V, Tedeschi E, Monticelli A, Siciliano A. Pleomorphic xanthoastrocytoma: Clinical, imaging and pathological features of four cases. Clin Neurol Neurosurg. 1997. 99: 40-5
7. Chang HT, Latorre JG, Hahn S, Dubowy R, Schelper RL. Pediatric cerebellar pleomorphic xanthoastrocytoma with anaplastic features: A case of long-term survival after multimodality therapy. Childs Nerv Syst. 2006. 22: 609-13
8. Edwards MS, Hudgins RJ, Wilson CB, Levin VA, Wara WM. Pineal region tumors in children. J Neurosurg. 1988. 68: 689-97
9. Ferrer E, Santamarta D, Garcia-Fructuoso G, Caral L, Rumia J. Neuroendoscopic management of pineal region tumours. Acta neurochir. 1997. 139: 12-20
10. Gangemi M, Maiuri F, Colella G, Buonamassa S. Endoscopic surgery for pineal region tumors. Minim Invasive Neurosurg. 2001. 44: 70-3
11. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B. Pleomorphic xanthoastrocytoma: What do we really know about it?. Cancer. 1999. 85: 2033-45
12. Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR. Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol. 2002. 26: 479-85
13. Gil-Gouveia R, Cristino N, Farias JP, Trindade A, Ruivo NS, Pimentel J. Pleomorphic xanthoastrocytoma of the cerebellum: Illustrated review. Acta Neurochir. 2004. 146: 1241-4
14. Hamlat A, Le Strat A, Guegan Y, Ben-Hassel M, Saikali S. Cerebellar pleomorphic xanthoastrocytoma: Case report and literature review. Surg Neurol. 2007. 68: 89-94
15. Herpers MJ, Freling G, Beuls EA. Pleomorphic xanthoastrocytoma in the spinal cord. Case report. J Neurosurg. 1994. 80: 564-9
16. Hirato J, Nakazato Y. Pathology of pineal region tumors. J Neurooncol. 2001. 54: 239-49
17. Hirose T, Ishizawa K, Sugiyama K, Kageji T, Ueki K, Kannuki S. Pleomorphic xanthoastrocytoma: A comparative pathological study between conventional and anaplastic types. Histopathology. 2008. 52: 183-93
18. Katayama K, Asano K, Shimamura N, Ogasawara Y, Naraoka M, Ohkuma H, Kurose A. A case of pleomorphic xanthoastrocytoma with anaplastic features in the pineal gland. Brain Tumor Pathol. 2013. 30: 242-6
19. Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: A distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979. 44: 1839-52
20. Kumar P, Tatke M, Sharma A, Singh D. Histological analysis of lesions of the pineal region: A retrospective study of 12 years. Pathol Res Pract. 2006. 202: 85-92
21. Kumar S, Retnam TM, Menon G, Nair S, Bhattacharya RN, Radhakrishnan VV. Cerebellar hemisphere, an uncommon location for pleomorphic xanthoastrocytoma and lipidized glioblastoma multiformis. Neurol India. 2003. 51: 246-7
22. Kurschel S, Lellouch-Tubiana A, Kulkarni AV, Sainte-Rose C. Pleomorphic xanthoastrocytoma of the cerebellopontine angle in a child. Childs Nerv Syst. 2006. 22: 1479-82
23. Morgenstern PF, Souweidane MM. Pineal region tumors: Simultaneous endoscopic third ventriculostomy and tumor biopsy. World Neurosurg. 2013. 79: S18.e9-13
24. Naidich MJ, Walker MT, Gottardi-Littell NR, Han G, Chandler JP. Cerebellar pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1. Neuroradiology. 2004. 46: 825-9
25. Nakamura M, Chiba K, Matsumoto M, Ikeda E, Toyama Y. Pleomorphic xanthoastrocytoma of the spinal cord. Case report. J Neurosurg Spine. 2006. 5: 72-5
26. Nitta J, Tada T, Kyoshima K, Goto T, Ishii K, Hongo K. Atypical pleomorphic astrocytoma in the pineal gland: Case report. Neurosurgery. 2001. 49: 1458-60
27. O’Brien DF, Hayhurst C, Pizer B, Mallucci CL. Outcomes in patients undergoing single-trajectory endoscopic third ventriculostomy and endoscopic biopsy for midline tumors presenting with obstructive hydrocephalus. J Neurosurg. 2006. 105: 219-26
28. Ohta T, Yachi K, Ogino A, Yokoyama T, Fukushima T, Watanabe T. Pleomorphic granular cell astrocytoma in the pineal gland: Case report. Neuropathology. 2010. 30: 615-20
29. Rosemberg S, Rotta JM, Yassuda A, Velasco O, Leite CC. Pleomorphic xanthoastrocytoma of the cerebellum. Clin Neuropathol. 2000. 19: 238-42
30. Saikali S, Le Strat A, Heckly A, Stock N, Scarabin JM, Hamlat A. Multicentric pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1. Case report and review of the literature. J Neurosurg. 2005. 102: 376-81
31. Simal-Julian JA, Sanchis-Martin R, Prat-Acin R, Miranda-Lloret P, Conde-Sardon R, Cardenas-Ruiz-Valdepenas E. Spinal pleomorphic xantoastrocytoma. Case report. Neurocirugia. 2010. 21: 390-5
32. Snipes GJ, Horoupian DS, Shuer LM, Silverberg GD. Pleomorphic granular cell astrocytoma of the pineal gland. Cancer. 1992. 70: 2159-65
33. Srinivas BH, Uppin MS, Panigrahi MK, Vijaya Saradhi M, Jyotsna Rani Y, Challa S. Pleomorphic xanthoastrocytoma of the pineal region. J Clin Neurosci. 2010. 17: 1439-41
34. Thakkar JP, Chew L, Villano JL. Primary CNS germ cell tumors: Current epidemiology and update on treatment. Med Oncol. 2013. 30: 496-
35. Vu TM, Liubinas SV, Gonzales M, Drummond KJ. Malignant potential of pleomorphic xanthoastrocytoma. J Clin Neurosci. 2012. 19: 12-20
36. Wasdahl DA, Scheithauer BW, Andrews BT, Jeffrey RA. Cerebellar pleomorphic xanthoastrocytoma: Case report. Neurosurgery. 1994. 35: 947-50
37. Yamini B, Refai D, Rubin CM, Frim DM. Initial endoscopic management of pineal region tumors and associated hydrocephalus: Clinical series and literature review. J Neurosurg. 2004. 100: 437-41
38. Yeaney GA, O’Connor SM, Jankowitz BT, Hamilton RL. A 16-year-old male with a cerebellar mass. Brain Pathol. 2009. 19: 167-70
39. Zarate JO, Sampaolesi R. Pleomorphic xanthoastrocytoma of the retina. Am J Surg Pathol. 1999. 23: 79-81
40. Zulch KJ. Reflections on the surgery of the pineal gland (a glimpse into the past). Gleanings from medical history. Neurosurg Rev. 1981. 4: 159-63

