

- Department of Neurosciences, University of the Philippines-Philippine General Hospital, College of Medicine and Philippine General Hospital, University of the Philippine, Manila, Philippines.
Correspondence Address:
Mary Angeline Luz Ubias Hernandez, Department of Neurosciences, University of the Philippines- Philippine General Hospital, College of Medicine and Philippine General Hospital University of the Philippine, Manila, Philippines.
DOI:10.25259/SNI_644_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Mary Angeline Luz Ubias Hernandez, Kathleen Joy O Khu. Calvarial metastasis from malignant pheochromocytoma associated with multiple endocrine neoplasia. 30-Sep-2022;13:444
How to cite this URL: Mary Angeline Luz Ubias Hernandez, Kathleen Joy O Khu. Calvarial metastasis from malignant pheochromocytoma associated with multiple endocrine neoplasia. 30-Sep-2022;13:444. Available from: https://surgicalneurologyint.com/surgicalint-articles/11906/
Date of Submission
18-Jul-2022
Date of Acceptance
06-Sep-2022
Date of Web Publication
30-Sep-2022
Abstract
Background: Malignant pheochromocytoma is a rare neuroendocrine tumor that may metastasize to the bones, liver, lungs, kidneys, and lymph nodes. Cerebral and skull metastases are even rarer, with only 17 cases reported in the literature. To the best of the authors’ knowledge, this is the first reported case of a purely calvarial metastasis from malignant pheochromocytoma associated with multiple endocrine neoplasia type 2A (MEN2A).
Case Description: A 31-year-old Filipino man diagnosed with MEN2A was found to have elevated urine metanephrine on routine surveillance, and workup revealed right adrenal and hepatic masses and a focus of intense tracer accumulation on the right frontal bone on metaiodobenzylguanidine I-123 scan. All the newly discovered lesions were resected to achieve tumor control. Histopathology revealed a diagnosis of pheochromocytoma for the calvarial lesion.
Conclusion: Malignant pheochromocytoma may give rise to indolent metastatic foci that can easily be missed without a thorough examination. Misdiagnosis and delays in management of this disease can be detrimental, resulting in irreversible complications and death.
Keywords: Calvarial metastasis, Malignant pheochromocytoma, Multiple endocrine neoplasia type 2A, Pheochromocytoma
Pheochromocytoma is a rare neuroendocrine tumor arising from chromaffin cells in the adrenal medulla.[
A 31-year-old Filipino man, previously diagnosed to have MEN 2A syndrome based on genetic testing and the presence of bilateral pheochromocytomas, medullary thyroid cancer, and parathyroid adenoma, was found to have elevated urine metanephrine on routine surveillance. He was clinically well, with an unremarkable physical and neurologic examination. Workup revealed right adrenal and hepatic masses, as well as a focus of intense tracer accumulation in the right frontal bone on MIBG-I-123 scan. Cranial CT scan showed a 2.3 × 2.3 × 0.7 cm well-defined, expansile, and lytic lesion with an enhancing soft-tissue component in the right frontal bone, with no apparent intracranial involvement [
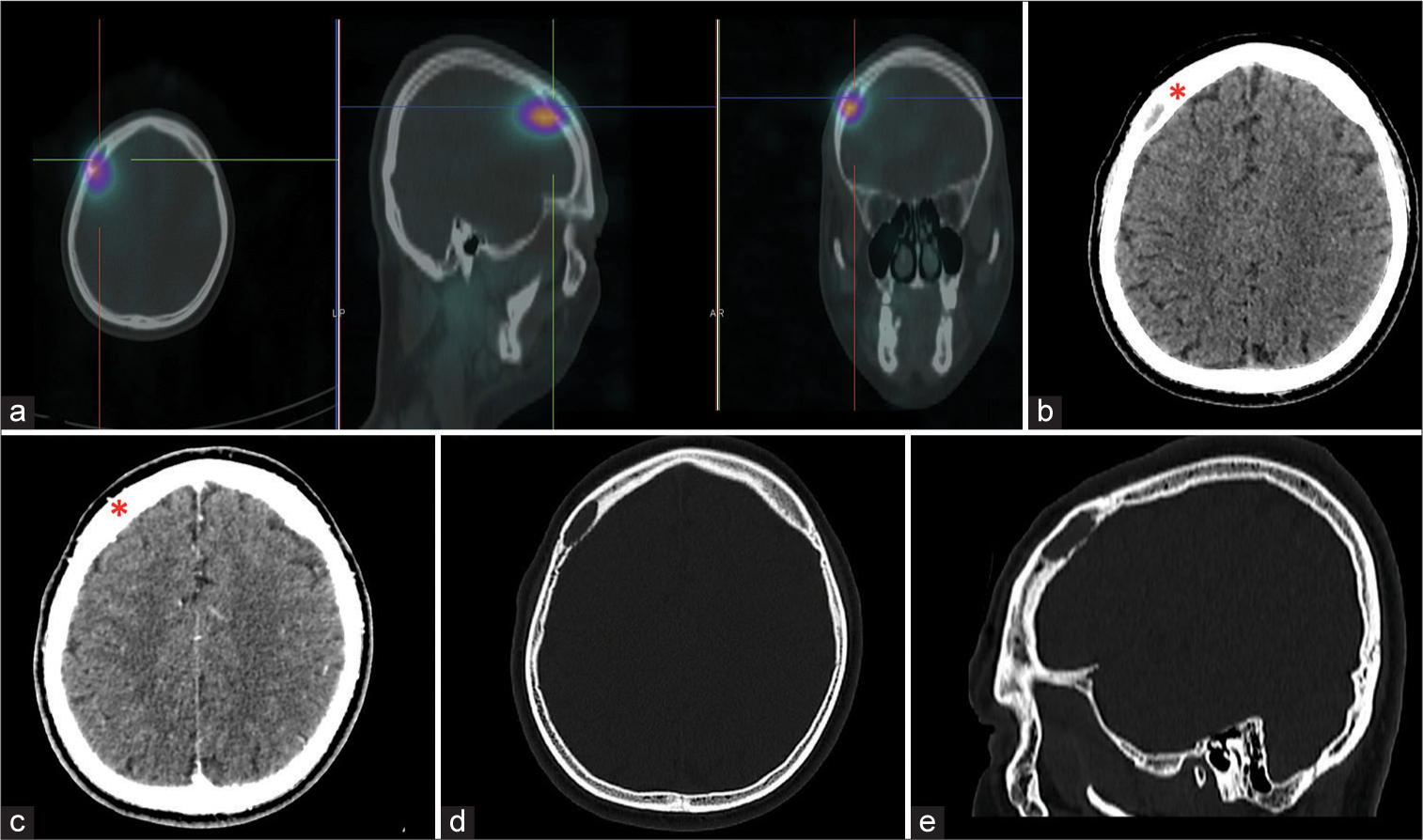
Figure 1:
(a) Axial, sagittal, and coronal images showing the focus of intense tracer accumulation in the right frontal bone on metaiodobenzylguanidine I-123 scintigraphy, (b) axial image of plain cranial computed tomography (CT) showing the well-defined, expansile, and elongated lytic lesion in the right frontal bone (red asterisk) measuring 2.3 × 2.3 × 0.7 cm, with enhancement on contrast (c), (d and e) axial and sagittal view of the cranial CT bone window showing the lytic lesion in the right frontal bone.
The patient underwent resection of the right adrenal and hepatic masses, followed by excision of the calvarial lesion. Intraoperatively, the outer and inner tables were found to be grossly intact. Histopathology revealed a diagnosis of pheochromocytoma for the right adrenal and calvarial lesions, and metastatic medullary thyroid carcinoma for the hepatic mass [

Figure 2:
Photomicrographs of the calvarial lesion. (a) Hematoxylin and eosin staining, 400×, showing polygonal cells with round to oval nuclei and abundant granular cytoplasm, immunohistochemical staining with chromogranin (b), and synaptophysin (c) were strongly and diffusely positive, consistent with pheochromocytoma.
Malignant pheochromocytomas may metastasize to nearby bony structures, liver, lungs, kidneys, and lymph nodes, but calvarial and intracranial metastases are less common.[
It has been postulated that pheochromocytoma metastasizes only to tissues with the same cell of origin.[
A complete surgical resection of the calvarial lesion was performed on our patient despite the absence of symptoms. The treatment of malignant pheochromocytoma involves resection of the primary tumor and removal of all foci of metastases to control the overproduction of catecholamines.[
This case demonstrated the importance of a thorough examination for patients with malignant pheochromocytoma. Unlike other foci, calvarial metastases may be indolent and asymptomatic, which may result in misdiagnosis and delay in management.
Statement of authorship
M.U.H: Conceptualization; resource; data curation; writing – original draft preparation; and writing – review and editing. K.O.K: Conceptualization; writing – original draft preparation; writing – review and editing; supervision; and project administration.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ajalle R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009. 41: 687-96
2. Chen JC, Zhuang DZ, Luo C, Chen WQ. Malignant pheochromocytoma with cerebral and skull metastasis: A case report and literature review. World J Clin Cases. 2021. 9: 2791-800
3. Chung KF, Sicard F, Vukicevic V, Hermann A, Storch A, Huttner WB. Isolation of neural crest derived chromaffin progenitors from adult adrenal medulla. Stem Cells. 2009. 27: 2602-13
4. Čtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018. 15: 3151-60
5. Cuerda C, Castejón I, Astigarraga B, Blanco C, Lucas T, Barceló B. A malignant extra-adrenal pheochromocytoma. Apropos 2 cases. Rev Clin Esp. 1993. 192: 76-8
6. Hashiba T, Maruno M, Fujimoto Y, Suzuki T, Wada K, Isaka T. Skull metastasis from papillary thyroid carcinoma accompanied by neurofibromatosis Type 1 and pheochromocytoma: Report of a case. Brain Tumor Pathol. 2006. 23: 97-100
7. Jandou I, Moataz A, Dakir M, Debbagh A, Aboutaieb R. Malignant pheochromocytoma: A diagnostic and therapeutic dilemma. Int J Surg Case Rep. 2021. 83: 106009
8. Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002. 241: 106-16
9. Muneer T, Tariq A, Siddiqui AH, Amanullah M. Malignant pheochromocytoma with widespread bony and pulmonary metastases. Cureus. 2018. 10: e3348
10. Pacak K, Ilias I, Adams KT, Eisenhofer G. Biochemical diagnosis, localization and management of pheochromocytoma: Focus on multiple endocrine neoplasia Type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J Intern Med. 2005. 257: 60-8
11. Yoshida S, Hatori M, Noshiro T, Kimura N, Kokubun S. Twenty-six-years’ survival with multiple bone metastasis of malignant pheochromocytoma. Arch Orthop Trauma Surg. 2001. 121: 598-600

