

- Department of Neurosurgery, Kyushu University,
- Department of Neurosurgery, Harasanshin Hospital,
- Clinical Chemistry and Laboratory Medicine, Kyushu University Hospital,
- Department of Pediatrics, Kyushu University,
- Department of Department of Otorhinolaryngology, Kyushu University.
Correspondence Address:
Takafumi Shimogawa
Department of Neurosurgery, Kyushu University,
DOI:10.25259/SNI_228_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Takafumi Shimogawa1, Nobutaka Mukae1, Takato Morioka2, Ayumi Sakata3, Yasunari Sakai4, Nozomu Matsumoto5, Masahiro Mizoguchi1. Corpus callosotomy for drug-resistant epilepsy in a pediatric patient with Waardenburg syndrome Type I. 10-May-2021;12:217
How to cite this URL: Takafumi Shimogawa1, Nobutaka Mukae1, Takato Morioka2, Ayumi Sakata3, Yasunari Sakai4, Nozomu Matsumoto5, Masahiro Mizoguchi1. Corpus callosotomy for drug-resistant epilepsy in a pediatric patient with Waardenburg syndrome Type I. 10-May-2021;12:217. Available from: https://surgicalneurologyint.com/surgicalint-articles/10797/
Date of Submission
06-Mar-2021
Date of Acceptance
14-Apr-2021
Date of Web Publication
10-May-2021
Abstract
Background: Waardenburg syndrome (WS) is caused by autosomal dominant mutations. Since the coexistence of epilepsy and WS type I is rare, the detailed clinical features and treatment of epilepsy, including surgery, have not been fully reported for these patients. We report the first case of an individual with WS type I, who underwent corpus callosotomy (CC) for drug-resistant epilepsy and obtained good seizure outcomes.
Case Description: A boy was diagnosed as having WS type I and developmental delay based on characteristic symptoms and a family history of hearing loss. He underwent cochlear implantation at 18 months of age. At 4 years of age, he developed epileptic seizures with a semiology of drop attack. Electroencephalography (EEG) showed bilateral synchronous high-amplitude spikes and wave bursts, dominant in the right hemisphere. Based on the multimodality examinations, we considered that ictal discharges propagated from the entire right hemisphere to the left, resulting in synchronous discharge and a clinical drop attack; therefore, CC was indicated. At 9 years of age, he underwent a front 2/3rd CC. At 1 year, the patient became seizure free, and interictal EEG showed less frequent and lower amplitude spike and wave bursts than before.
Conclusion: When patients with WS Type I and cognitive impairment show drug-resistant epilepsy, clinicians should consider a presurgical evaluation.
Keywords: Corpus callosotomy, Drug-resistant epilepsy, Genetic dysmorphic syndrome, Waardenburg syndrome
INTRODUCTION
Waardenburg syndrome (WS) is caused by autosomal dominant mutations with high penetrance. Identified for the 1st time in 1947,[
CASE REPORT
A boy weighing 3126 g at birth, with Apgar scores of 9 and 9, showed total heterochromia in the left eye, dystopia canthorum (W-index: 2.36),[
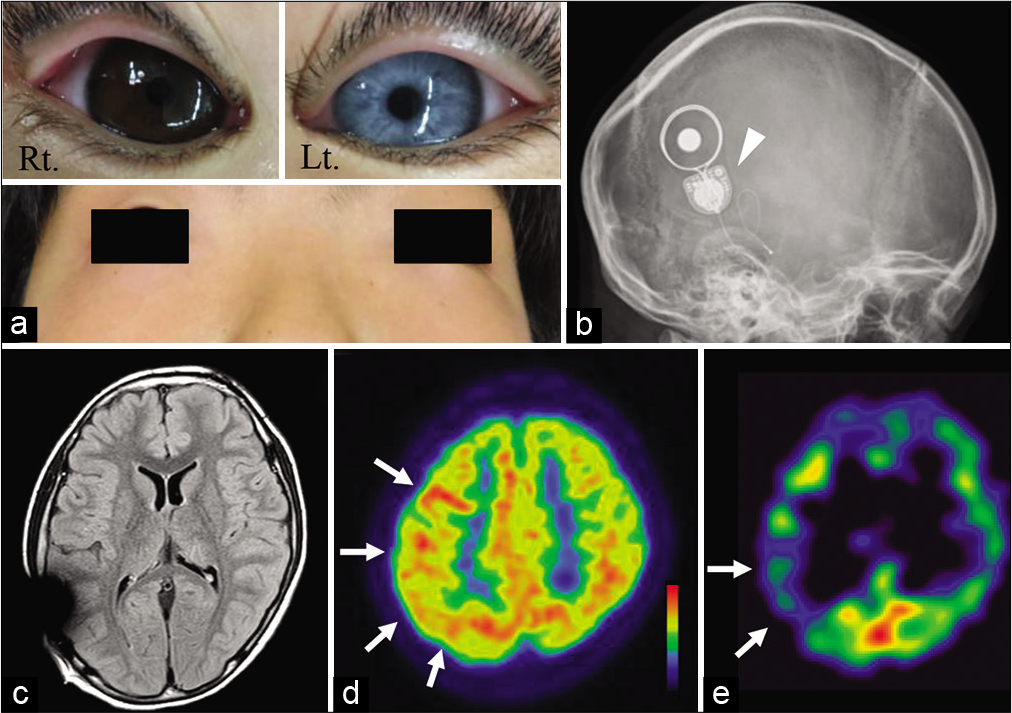
Figure 1:
(a) Photograph showing total heterochromia in the left eye, dystopia canthorum, and broad nasal root. (b) Plain head radiograph demonstrating the cochlear implant device for congenital sensorineural hearing loss (arrowhead). (c) Magnetic resonance images with a fluid-attenuated inversion recovery sequence revealing no obvious abnormal findings. (d) 18F-fluorodeoxyglucose-positron emission tomography depicting hypermetabolism in the right frontal lobe and parietal lobe (white arrows). (e) (123)I-iomazenil (IMZ) single-photon emission computed tomography showing decreased IMZ uptake in the right parietal lobe in a late image (white arrows).
At 4 years of age, he developed epileptic seizures with a semiology of drop attack, left upper limb atony, and left eyelid myoclonia, followed by focal to bilateral tonic–clonic seizures (FBTCS). He was diagnosed with epilepsy, and antiepileptic drug (AED) administration initiated. However, despite treatment with optimal AED dosages, including levetiracetam (LEV), valproate (VPA), perampanel (PRP), clobazam (CLB), ethosuximide (ESM), lacosamide, and zonisamide, the intractable seizures persisted.
He was referred to us at 9 years of age. He had severe cognitive impairment (intelligence quotient of 33) and behavioral features of autism spectrum disorder. His epileptic seizures included daily multiple drop attacks, and weekly left eyelid myoclonia followed by FBTCS, despite treatment with multiple AEDs including 1000 mg of LEV, 500 mg of VPA, 4 mg of PRP, 10 mg of CLB, and 500 mg of ESM. His seizures were characterized by repeated appearance and disappearance; if present, they persisted for approximately 2 weeks. Further, following a drop attack, he also experienced a head trauma.
Magnetic resonance imaging (MRI) performed after removal of the cochlear implant magnet showed no obvious abnormal findings (he underwent replacement of cochlear magnetic implant after the MRI) [
On video electroencephalography (EEG), ictal EEG showed bilaterally synchronous high-amplitude spikes and wave bursts, dominant in the right hemisphere. Six seconds after spike burst appearance, the EEG exhibited a mild attenuation when the patient experienced a drop attack [
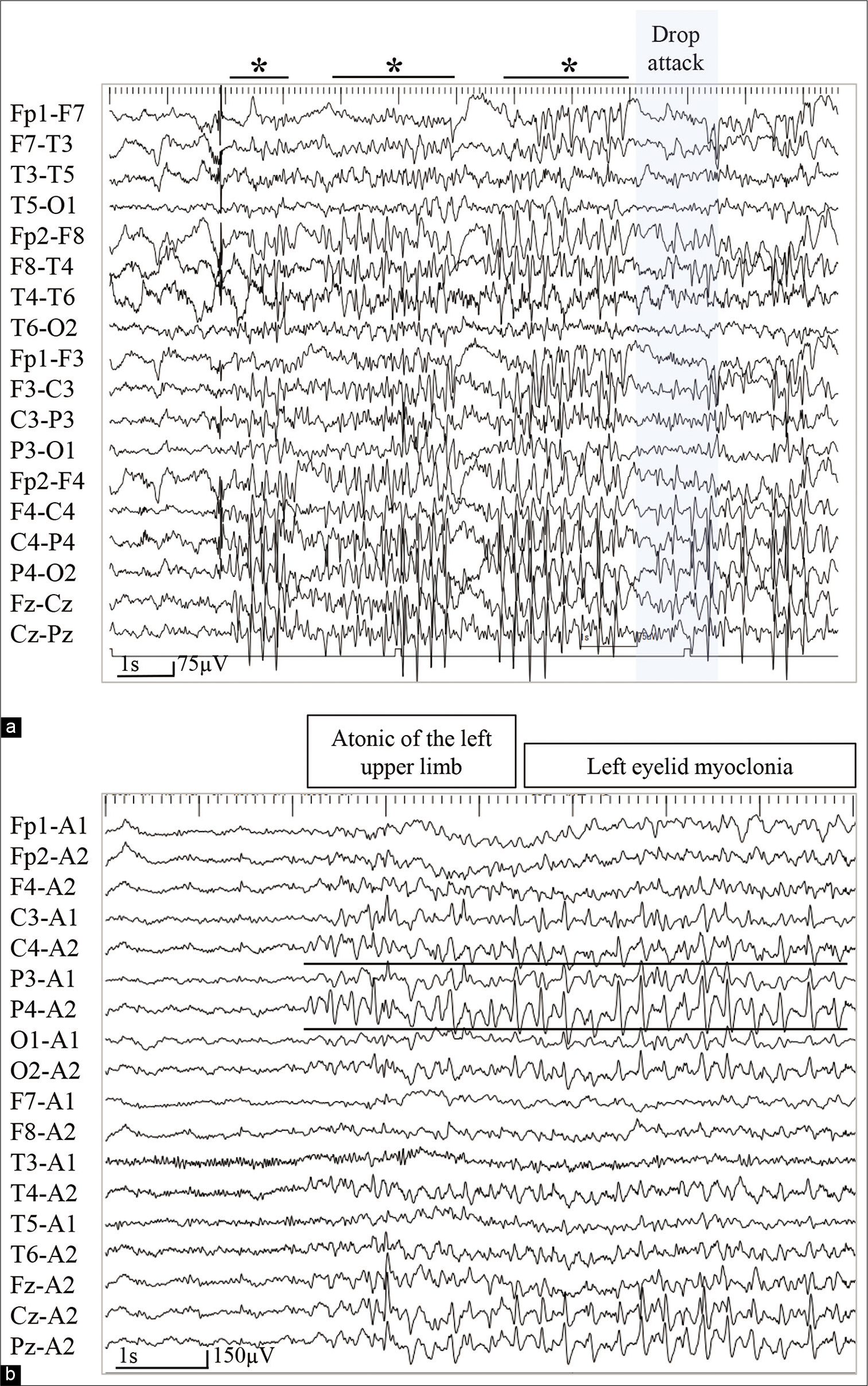
Figure 2:
(a) On video electroencephalogram (EEG), ictal EEG showing bilaterally synchronous high-amplitude spike and wave bursts, right hemisphere dominant (Asterisks). Six seconds after the appearance of spike bursts, the EEG shows a mild attenuation when the patient shows a drop attack (blue box). (b) Another ictal EEG revealing background activity attenuation, followed by continuous poly spike-and-wave activities from the parieto-occipital region at C4 and P4 of the International 10–20 EEG system (black lines). The patient experienced an atonic seizure of the left upper limb and left eyelid myoclonia during epileptiform discharges.
From multimodal examinations, we considered that ictal discharges originating from the entire right hemisphere abruptly propagated to the left hemisphere, resulting in synchronous discharge on both sides and a clinical drop attack; thus, a Stage II evaluation placing a chronic subdural electrode on the entire right hemisphere was deemed complicated, while placing a chronic subdural electrode was considered too invasive for a 9-year-old boy with severe cognitive impairment. Therefore, we focused on the drop attack as main symptom and decided to perform a CC.
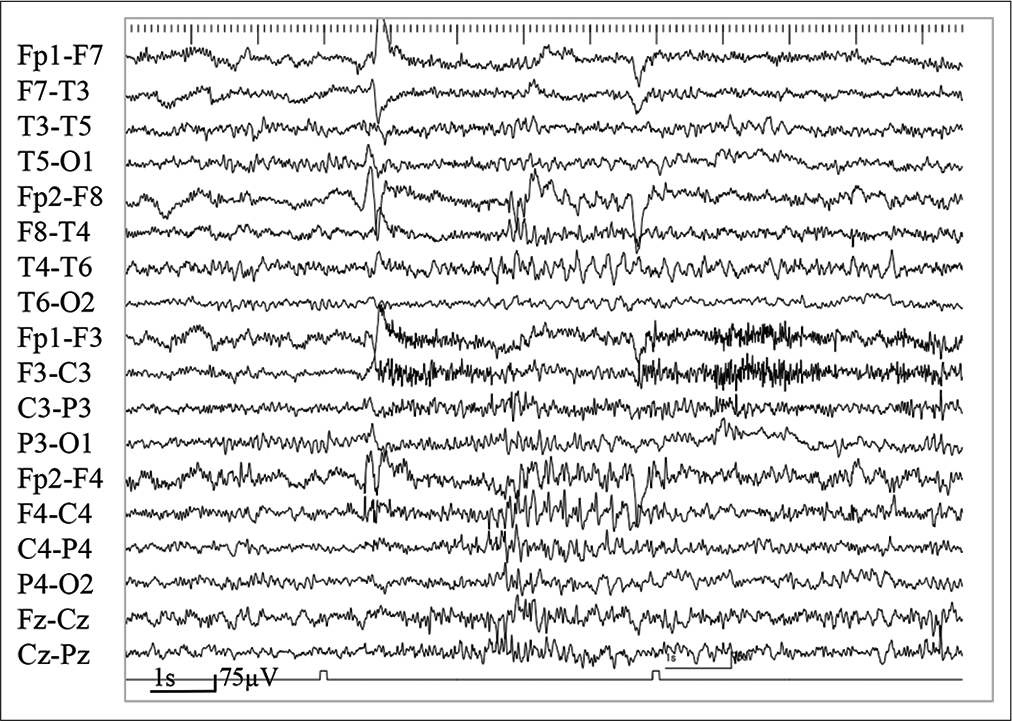
The patient underwent a front 2/3 CC. The cochlear implant magnet was removed again and replaced by a nonmagnetic titanium plug by an otolaryngologist just before the CC, and a new magnet was inserted after the postoperative MRI was performed the next day. After CC, the frequency of drop attacks gradually decreased. Seven months after the CC, the drop attack disappeared. One year after CC, the patient became seizure free. Interictal EEG at 1 year after CC showed less frequent and lower amplitude asynchronous spike and wave bursts, right hemisphere dominant, than before surgery [
DISCUSSION
WS has been reported to be caused by mutations in six genes: PAX3, MITF, EDN3, EDNRB, SOX10, and SNAI2,[
Epilepsy is one of the most common findings in genetic dysmorphic syndromes and more than one-third of these patients have drug-resistant epilepsy.[

In the present case, seizure became drug resistant. We considered that, with multimodal examination, ictal discharges originating from the right entire hemisphere propagated to the left, resulting in synchronous discharge and a clinical drop attack, leading to an indication for CC. This hypothesis was later supported by the postoperative seizure outcomes and EEG findings. Here, we describe the first case of a WS Type I patient who underwent CC for drug-resistant epilepsy and achieved seizure freedom. To the best of our knowledge, this is the first case of CC in a cochlear implant recipient. If a patient is a cochlear implant recipient, such as in the present case, a multidisciplinary team of neurosurgeons, otolaryngologists, and speech-language hearing therapists is necessary to ensure the patient receives maximum benefit from the intervention.
CONCLUSION
When patients with WS Type I and cognitive impairment show drug-resistant epilepsy, clinicians should consider a preoperative evaluation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JP19K24314 to T.S.).
Conflicts of interest
There are no conflicts of interest.
References
1. Alfei E, Raviglione F, Franceschetti S, D’Arrigo S, Milani D, Selicorni A. Seizures and EEG features in 74 patients with genetic-dysmorphic syndromes. Am J Med Genet A. 2014. 164A: 3154-61
2. Bondurand N, Dastot-Le F, Stanchina L, Collot N, Baral V, Marlin S. Deletions at the SOX10 gene locus cause Waardenburg syndrome Types 2 and 4. Am J Hum Genet. 2007. 81: 1169-85
3. Cantani A, Bamonte G, Tacconi ML. Mental retardation and EEG abnormalities in Waardenburg’s syndrome: Two case reports (EEG anomalies in Waardenburg’s syndrome). Pediatr Padol. 1989. 24: 137-40
4. Chaoui A, Watanabe Y, Touraine R, Baral V, Goossens M, Pingault V. Identification and functional analysis of SOX10 missense mutation in different subtypes of Waardenburg syndrome. Hum Mutat. 2011. 32: 1436-49
5. Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH, Beighton P. Waardenburg syndrome (WS) Type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am J Hum Genet. 1992. 50: 902-13
6. Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004. 36: 361-9
7. Liu XZ, Newton VE, Read AP. Waardenburg syndrome Type II: Phenotypic findings and diagnostic criteria. Am J Med Genet. 1995. 55: 95-100
8. Newton VE. Waardenburg’s syndrome: A comparison of biometric indices used to diagnose lateral displacement of the inner canthi. Scand Audiol. 1989. 18: 221-3
9. Pantke OA, Cohen MM. The Waardenburg syndrome. Birth Defects Orig Artic Ser. 1971. 7: 147-52
10. Parington MW. Waardenburg’s syndrome and heterochromia iridum in a deaf school population. Can Med Assoc J. 1964. 90: 1008-17
11. Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997. 34: 656-65
12. Suzuki N, Mutai H, Miya F, Tsunoda T, Terashima H, Morimoto N. A case report of reversible generalized seizures in a patient with Waardenburg syndrome associated with a novel nonsense mutation in the penultimate exon of SOX10. BMC Pediatr. 2018. 18: 171
13. Sznajer Y, Coldéa C, Meire F, Delpierre I, Sekhara T, Touraine RL. A de novo SOX10 mutation causing severe Type 4 Waardenburg syndrome without Hirschsprung disease. Am J Med Genet A. 2008. 146: 1038-41
14. Warrdenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951. 3: 195-253
15. Warrdenburg PJ. Dystopia punctorum lacrimarum, blepharophimosis and partial iris atrophy at a deaf mute. Ned Tijdschr Geneeskd. 1948. 92: 3463-6

