

- Department of Neurosurgery, Lokmanya Tilak Municipal Medical College and Hospital, Mumbai, Maharashtra, India.
- Department of Neurosurgery, All India Institute of Medical Sciences, New Delhi, India.
Correspondence Address:
Subhasish Dey, Department of Neurosurgery, All India Institute of Medical Sciences, New Delhi, India.
DOI:10.25259/SNI_1218_2021
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Batuk Diyora1, Subhasish Dey2, Ashish Dubey1, Lavina Lakdawala1. Cranial fibrous dysplasia: An institutional experience and review of the literature. 25-Feb-2022;13:66
How to cite this URL: Batuk Diyora1, Subhasish Dey2, Ashish Dubey1, Lavina Lakdawala1. Cranial fibrous dysplasia: An institutional experience and review of the literature. 25-Feb-2022;13:66. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=11415
Date of Submission
08-Dec-2021
Date of Acceptance
18-Jan-2022
Date of Web Publication
25-Feb-2022
Abstract
Background: Cranial or craniofacial fibrous dysplasia (CFD) is a rare entity which most often presents with either incidental finding or with pain/cosmetic disfigurement or visual/hearing problems. Multidisciplinary treatment with close follow-up or medical management/surgery is options. Management of these lesions can often give satisfying results. There is a dearth of neurosurgical literature on this subject matter. Our objective was to review the clinical symptomatology and outcome of CFD patients managed in our institution.
Methods: This is a retrospective observational study of CFD patients managed in our institution over a period of 5 years. Clinical and radiological data were collected from departmental database. Outcomes were evaluated immediately and on 1–4 years follow-up.
Results: A total of 21 patients were managed over a period of 5 years with age ranging from 12 to 55 years and symptoms of cosmetic issues or visual disturbance. Preoperative computed tomography scan with 3D reconstruction with bone window was done in all patients. In most of the patients (16/20), immediate reconstruction was done following excision. Five patients were managed conservatively. Follow-up was obtained over a period ranging from 1 to 4 years and all (except one) patients are doing well. Only one patient had permanent visual impairment in spite of early intervention.
Conclusion: Craniofacial dysplasia has various modalities of management. Careful selection of patients for surgical or conservative management is feasible with good results both in short and long term.
Keywords: Bisphosphonates, Cranial fibrous dysplasia, Single-stage surgery, Visual outcome
INTRODUCTION
Fibrous dysplasia (FD) is a condition where normal bone is replaced with woven bone with interposed fibrous tissue in between [
FD is characterized by bony lesions and may be associated with endocrine and biochemical abnormality. Genetic mutation (GNAS1 gene) is linked up with this disease.[
Craniofacial bones are common locations of FDs mostly PFDs. They clinically present with cosmetic deformity, orbitocranial swelling, and dental malocclusion mostly. There can be associated pain, visual disturbance, hearing loss, nasal obstruction, and diplopia depending on the extent of involvement. FDs mostly develop and progress in the younger age group. Although MFD is less aggressive, slow progression even “burn out” up to adolescents, PFDs continue to progress over time and more symptomatic. On computed tomography (CT) scan, it has a ground-glass appearance with sclerotic/cystic/mixed pattern. Although conservative medical management is adopted in most nonprogressive asymptomatic cases patients, surgical resection with reconstruction whenever feasible and indicated is recommended and definitive for symptomatic cases.[
We present our experience of 21 such cases that were managed over 5 years along with short- and long-term outcomes.
MATERIALS AND METHODS
This is a retrospective study. Approval was taken from the Institutional Ethics Board. Data were collected for 21 symptomatic FD patients managed between 2014 and 2019. All endocrine and biochemical abnormalities were identified and corrected.
Inclusion criteria for surgical management:
The following criteria were included in the study:
Patient with cosmetic deformity wanting surgical treatment Functional symptoms such as diminution of vision, diplopia, and hearing loss.
Inclusion criteria for medical management or observation:
The following criteria were included in the study:
Mild-to-moderate pain with slowly progressive disease or asymptomatic case Lesions that are surgically not feasible to access Patient not willing for surgery.
Preoperative symptoms were analyzed and appropriate indications were determined. All patients underwent preoperative craniofacial CT scan with three-dimensional reconstructions with bone windows.
In 16 out of 21 patients who were managed surgically, a standard bicoronal incision was taken in most patients and flap was raised. The underlying bony abnormality was defined all around and the entire abnormal areas were excised in the form of drilling/curettage en bloc or piecemeal. The defect was delineated all around and immediate reconstruction was performed in all cases. Reconstruction was done with split-thickness bone graft alone or in combination with polymethylmethacrylate (PMMA) cement or titanium mesh [
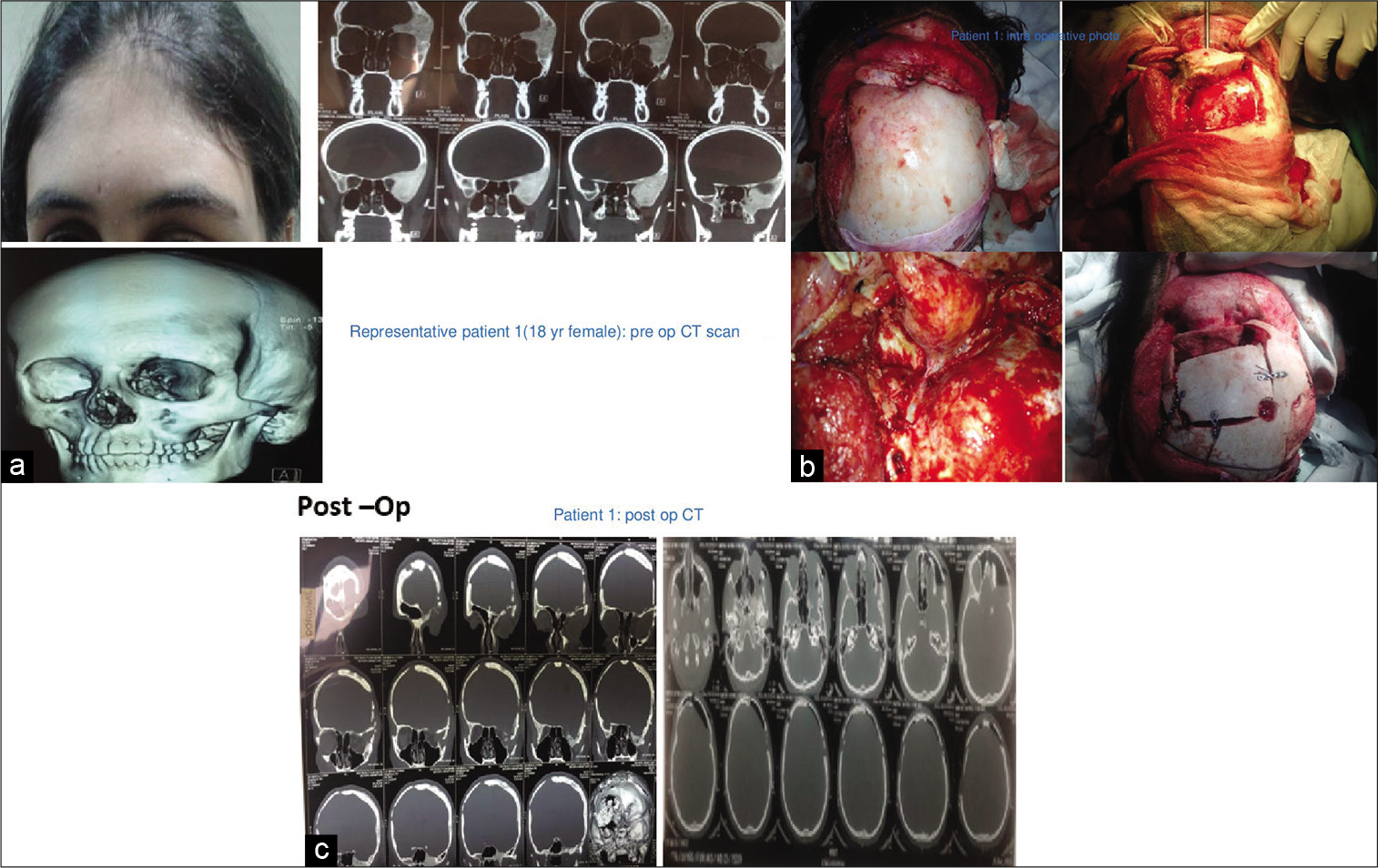
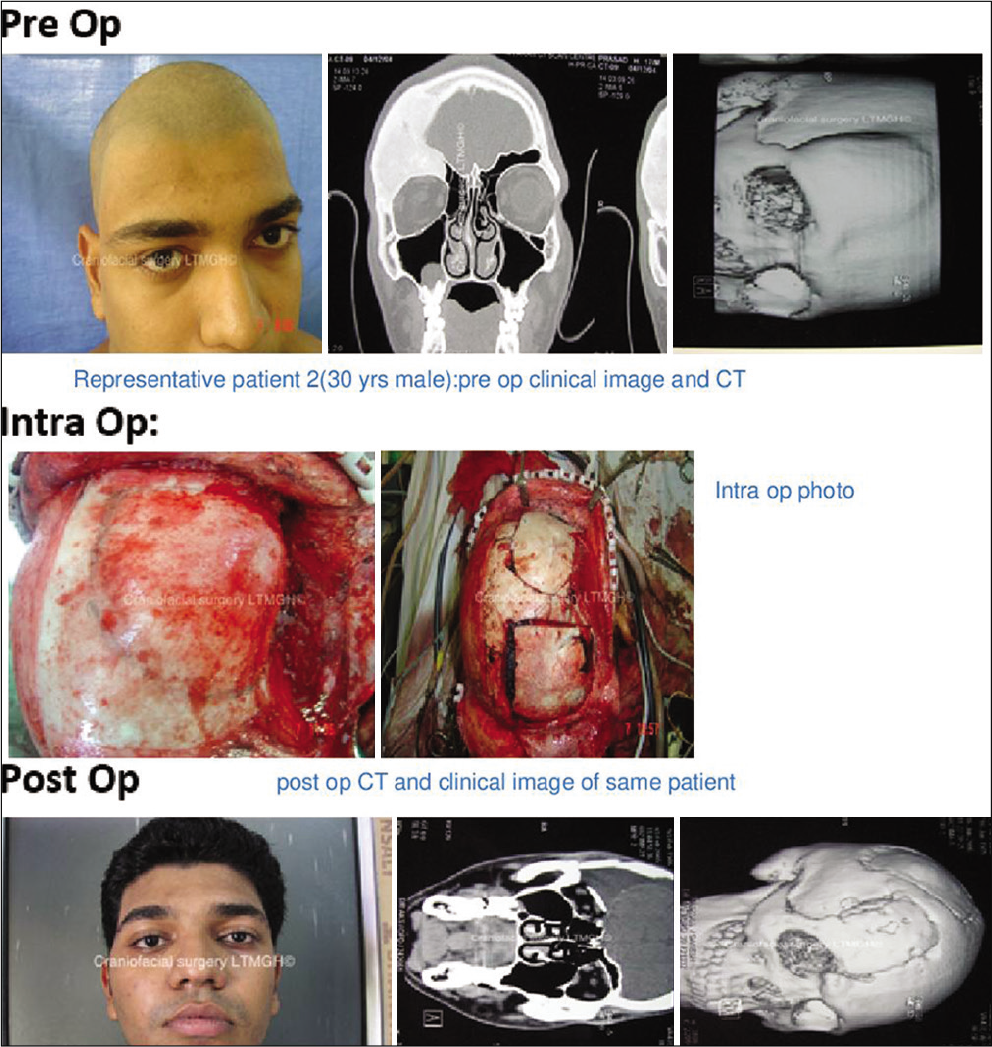
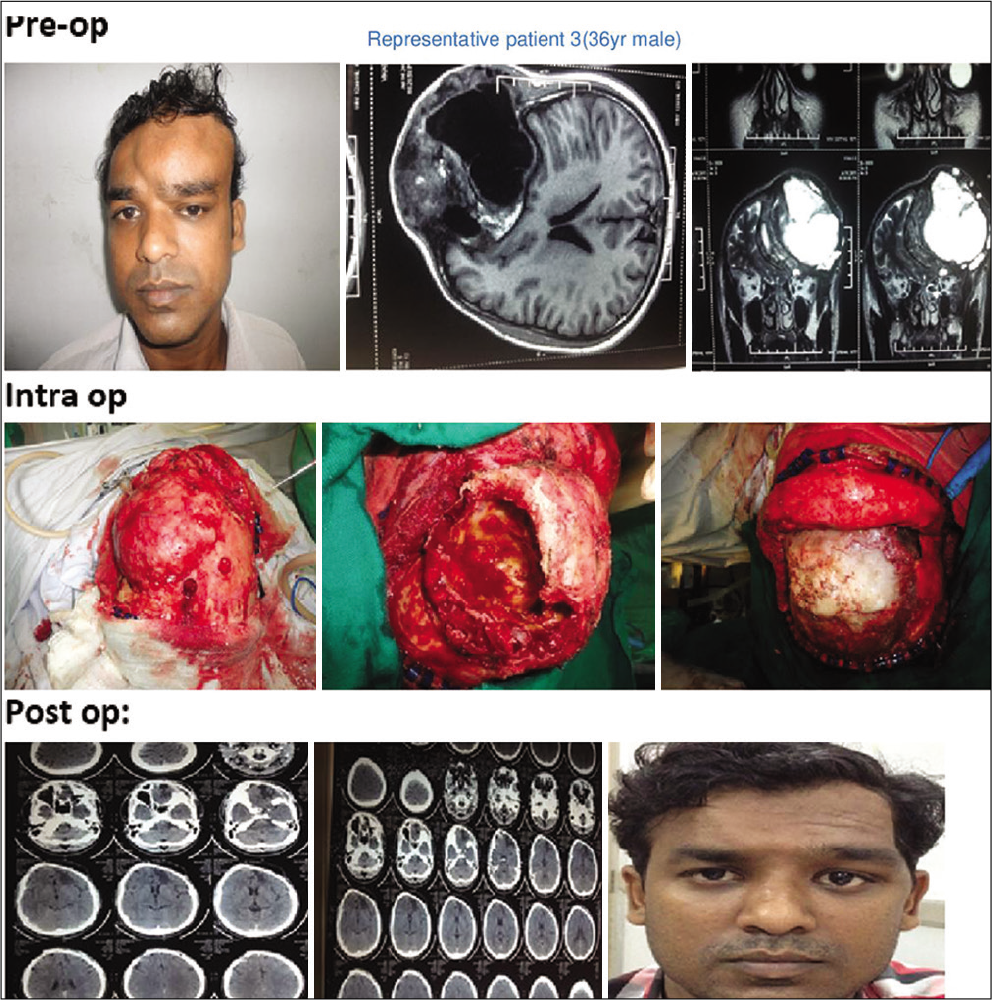
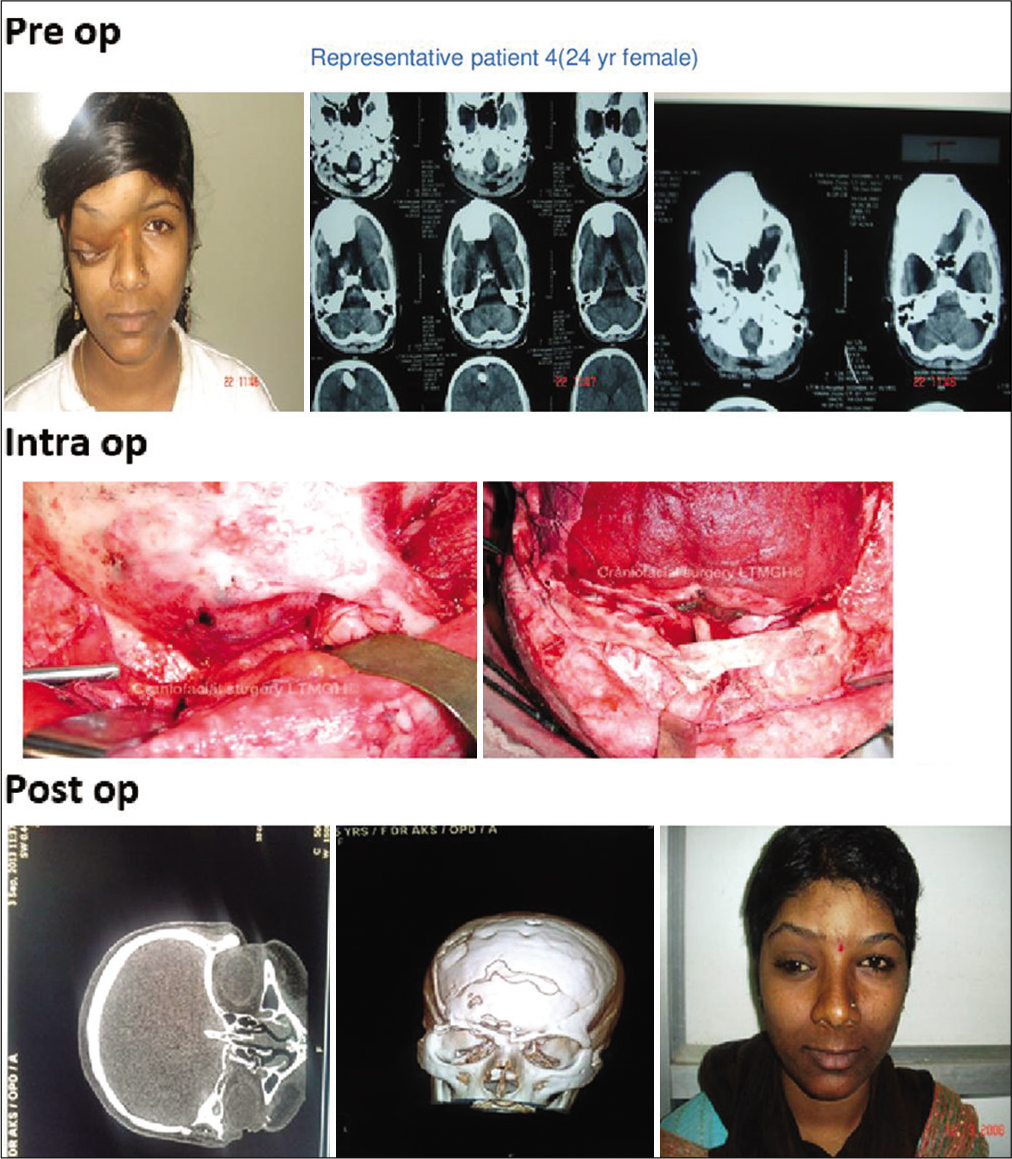
In patients who were managed medically for pain (3/21), intravenous pamidronate (bisphosphonates) was given 1–3 mg/kg every 6 months–1 yearly. Vitamin D supplements were also given.
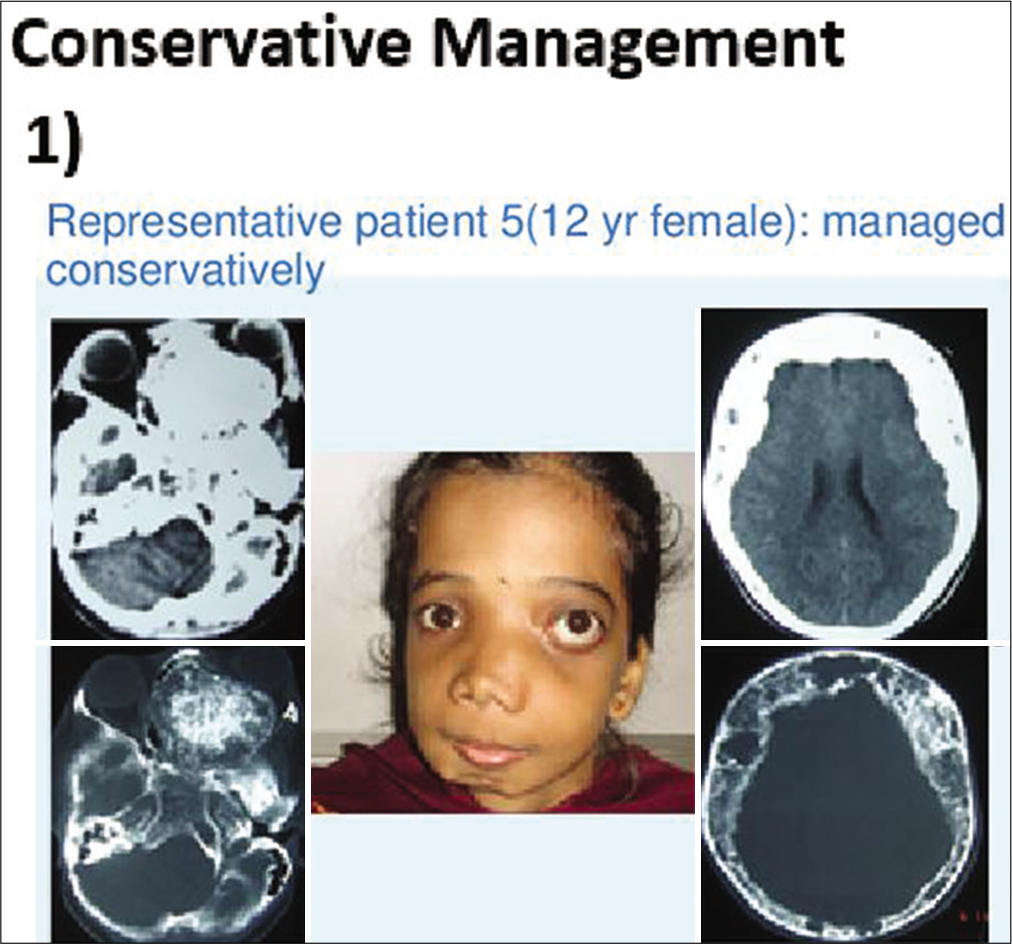
Two patients with holocranial/bilateral FD were managed with observation only. Clinical outcomes were assessed immediately as well as in short- and long-term follow-up over 1–4 years in both groups of patients [

RESULTS
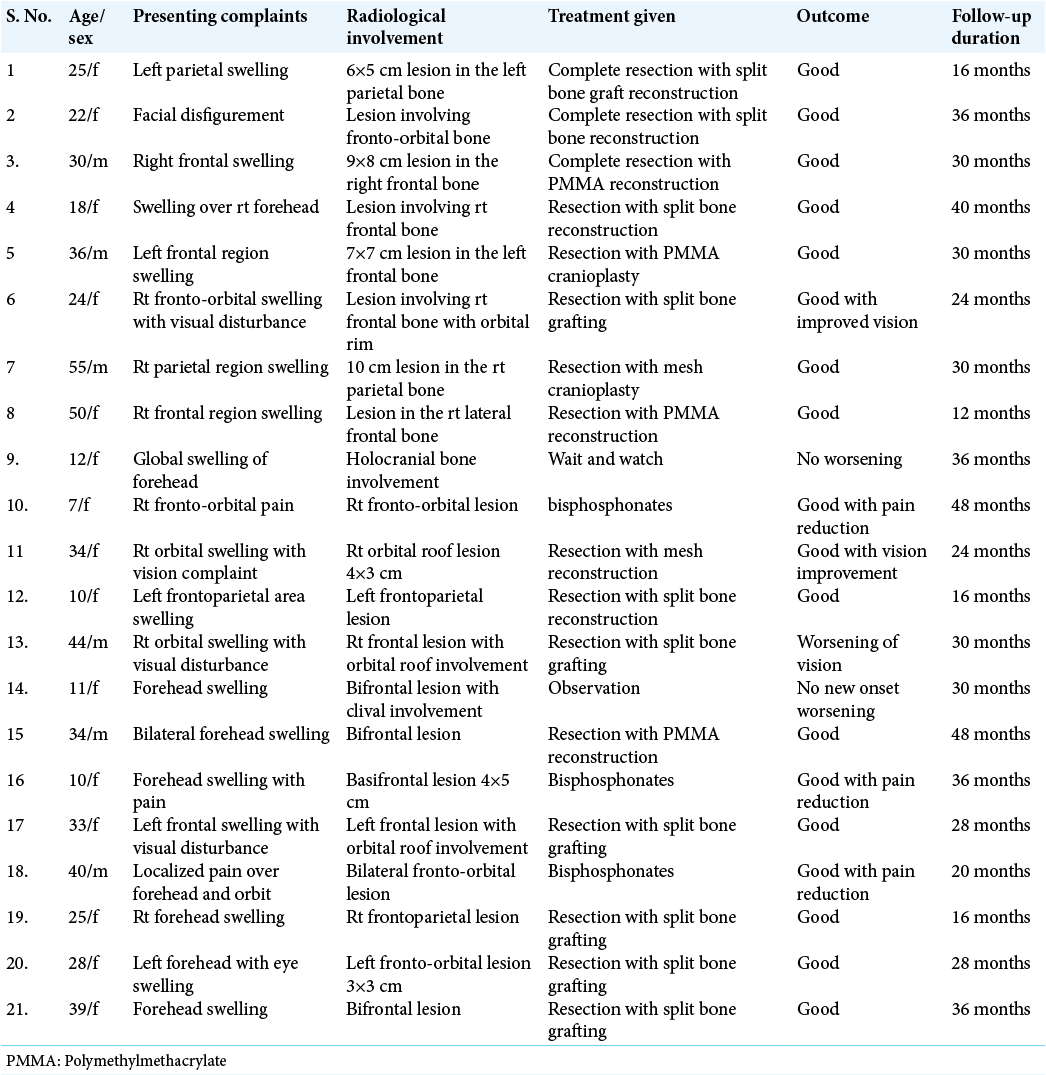
A total of 21 patients were treated over 5 years. Among them, 6 (29%) were male and 15 (71%) females. The age group ranges from 12 to 55 years with a median age of 26 years [

All patients underwent cranio-facio-orbital CT scan with three-dimensional reconstructions with bone windows for ruling out other differential diagnoses (e.g., metastasis, meningioma-related hyperostosis, and bony tumors) and for surgical planning in indicated patients [
In the surgical group following surgical excision of the lesion, immediate reconstruction was done. Split-thickness bone graft alone was used in 10 cases out of 16 (63%) [
In medically managed patients after ruling out other endocrine (parathyroid hormone, calcitonin, etc.) and biochemical (serum calcium, serum phosphate, etc.) abnormalities, intravenous pamidronate (1–3 mg/kg) 6 months–1 yearly until pain subsides along with Vitamin D and oral calcium supplements was given. Regular follow-up was done thereafter.
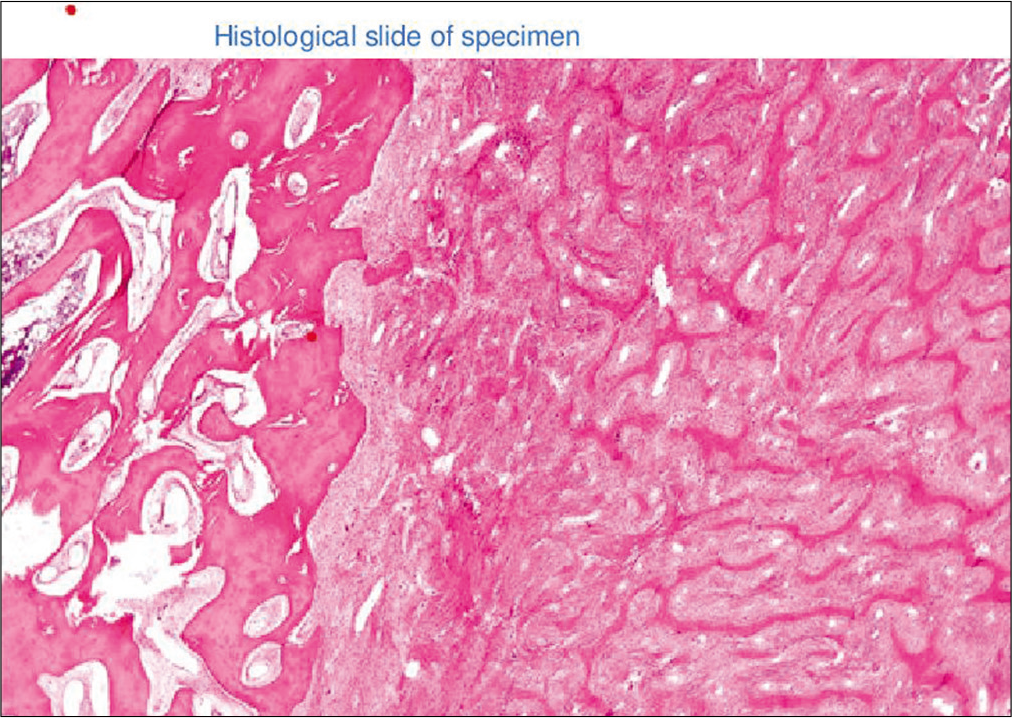
Histopathological analysis was done in operative samples and all cases showed a varying amount of spindle cells and trabeculae of immature woven bone [
Complications and follow-up
In surgically treated patients, one patient had persistent subgaleal collection which was managed by compression bandage and anti-inflammatory medication. Another patient experienced postoperative new-onset seizure that was managed by anti-seizure drugs.
Surgically managed patients were followed up over a period ranging from 12 to 48 months with an average follow-up period of 24–30 months [
In conservatively managed group, patients remained symptom free and nonprogressive disease over 18–24 months follow-up.
DISCUSSION
FD is a relatively uncommon condition and there is a relative dearth of studies on this topic in the neurosurgical literature. Its treatment is also controversial with many in favor of expectant/medical management. Surgical treatment whenever done can be accompanied by a staged reconstruction later or immediately at the time of first surgery. We here have presented our experience of single-stage surgery of these lesions in 16 patients as well as conservative treatment in appropriate cases.
FD is benign pathology of bones defined by Reed as an “arrest of bone maturation in woven bone with ossification resulting from metaplasia of a nonspecific fibro-osseous type”. The term FD was coined by Linchiestien and Jaffe (1942). FD is classified as monostotic type (MFD) (involving single bone) and polyostotic type (PFD) (multiple bones). Monostotic type (60–70%) is more common and commonly asymptomatic seen in early adulthood.[
Appendicular bones (tibia and femur), ribs, and craniofacial bones are more commonly involved. PFD (50–70%) is the most common presentation in craniofacial involvement. MFD comprises 10–30%. FD incidence is almost the same in both genders with slight female predilection. FD presents in late 20s and 30s after puberty when bone maturity occurs. It involved membranous bone more readily than cartilaginous bones. Malignant transformation occurs in <1% of cases.[
Craniofacial FD (CFD) usually presents with multiple adjacent skull and face bones; however, other body skeletons are not involved generally.[
Eversole et al.[ CFD, in which only the bones of the craniofacial complex are affected Lichtenstein-Jaffe type, in which multiple bones of the skeleton are involved with café-au-lait pigmentations on the skin and rare endocrinopathies in a few of these patients; and Albright’s syndrome, characterized by the triad of polyostotic FD (mostly unilateral), café-au-lait pigmentations on the skin, and various endocrinopathies. Another very rare and special form is the Mazabraud syndrome, describing an association of the FD with soft-tissue myxomas.
Etiology
Genetic changes lead to FD. Culprit mutation (missense) affecting the GNAS1 gene located at chromosome 20q13 has been implicated. Persistent activation of adenyl cyclase and increase cellular cAMP due to mutation causes abnormal bone formation and resorption of normal bone by increasing interleukin (IL)-6.[
Diagnosis
CFD generally presents with painless/painful bony enlargement with facial/cranial asymmetry. Involvement of orbit, skull base, neural foramina, and calvaria leads to seizures, visual diminution, diplopia, proptosis, hearing loss, etc.[
Differential diagnosis includes various skull tumor/lesions such as simple bone cyst, giants cell tumor, fibroxanthoma, osteofibroma, osteoma, and Paget disease for MCFD and eosinophilic granuloma, enchondromatosis, and brown tumor of hyperparathyroidism for PCFDs.[
Patients’ age, sex, and radiological features (X-ray, CT, and MRI) were assessed. A thin bony cortex was seen with well-defined borders and ground-glass appearance. Three distinctive patterns on CT were seen – (1) Scattered lesion with bony expansion (Pagetoid), (2) homogeneous with ground-glass appearance (sclerotic), and (3) low density with sclerotic margin (cystic).[
Histopathological diagnosis is confirmatory and useful to rule out any malignancy.
Management
CFD management includes conservative approach with observation and follow-up, medical management, and surgical including surgical remolding (conservative), radical surgery, and complete excision.
Small asymptomatic cosmetically acceptable lesions are best managed with observation only. Surgically difficult accessible sites of central skull base such as clivus, sphenoid, and petrous temporal bones (ZONE 3 Chen YR, Noordhoff, 1990) are advised to be managed with observation only as surgery can cause more harm [
Medical treatment, still not promising in the management of FDs and still under evaluation, is based on control of pathophysiology of abnormal bone formation and raised osteoclastic activity and decreasing local pain due to this. This includes bisphosphonates, more appropriately pamidronate intravenous 60 mg/day for 3 days repeated in every 6 months–1 year. Calcitonin is another drug used by some clinicians. Vitamin D and calcium supplements also used in selected patients.[
Surgery is the mainstay treatment in selected patients with CFDs. Radical surgery with excision is one option not feasible many times due to diffuse involvement and adjacent neurovascular structure.[
Conservative surgery including bone remolding and recontouring is performed most of the times to correct cosmetic deformities and functional deficit due to neural foramina or orbital involvement. The risk of recurrence (15– 20%) and less likely malignant transformation (0.4–4%) is possible in conservative approach which can be minimized by performing primary surgery after pubertal growth and bone maturity and regular follow-up by clinical, radiological (CT/ MRI/X-ray), and biochemical (serum alkaline phosphatase is raised in recurrence) examination.[
Couldwell et al.[
Lei et al.[
Adada and Al-Mefty[
Valentini et al.[
Lee et al.[
Zanotti et al.[
Limitations
The retrospective nature of this study is a limiting factor. Long follow-up has been done to compensate for the nature of the study. Another factor is the number of patients. However, given the dearth of adequate neurosurgical literature on this subject matter, our number is respectable.
CONCLUSION
CFD is complex lesions which range from asymptomatic (nonprogressive) to cosmetic disturbances, pain, neurological, ophthalmological, ENT, and dental symptoms in progressive disease. Age and symptoms should be considered before deciding management; however, single-stage surgical management of symptomatic CFD with immediate reconstruction in selected patients is feasible and safe with good short- and long-term outcomes. However, given the variable natural history of the disease, regular follow-up and long-term surveillance are needed.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Ethical approval
All procedures performed in this study were per with the ethical standards of the Institutional Research Committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
References
1. Adada B, Al-Mefty O. Fibrous dysplasia of the clivus. Neurosurgery. 2003. 52: 318-23
2. Alvares LC, Capelozza AL, Cardoso CL, Lima MC, Fleury RN, Damante JH. Monostotic fibrous dysplasia: A 23-year follow-up of a patient with spontaneous bone remodeling. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009. 107: 229-34
3. Ben Hadj Hamida F, Jlaiel R, Ben Rayana N, Mahjoub H, Mellouli T, Ghorbel M. Craniofacial fibrous dysplasia: A case report. J Fr Ophtalmol. 2005. 28: e6
4. Chen YR, Noordhoff MS. Treatment of craniomaxillofacial fibrous dysplasia: How early and how extensive?. Plast Reconstr Surg. 1990. 86: 835-42
5. Couldwell WT, Taussky P, Bowers CA. Surgical treatment of craniofacial fibrous dysplasia in adults. Neurosurg Rev. 2014. 37: 47-53
6. Erzurumlu ZU, Celenk P, Bulut E, Baris YS. CT imaging of craniofacial fibrous dysplasia. Case Rep Dent. 2015. 2015: 134123
7. Eversole LR, Sabes WR, Rovin S. Fibrous dysplasia: A nosologic problem in the diagnosis of fibro osseous lesions of the jaws. J Oral Pathol. 1972. 1: 189
8. Feingold RS, Argamaso RV, Strauch B. Free fibula flap mandible reconstruction for oral obstruction secondary to giant fibrous dysplasia. Plast Reconstr Surg. 1996. 97: 196-201
9. Ganau M, Paris M, Uff C. Images in Neuroscience: Calvarial hyperostosis associated with multiple intracranial tumours. J Clin Neurosci. 2018. 50: 029
10. Kim DD, Ghali GE, Wright JM, Edwards SP. Surgical treatment of giant fibrous dysplasia of the mandible with concomitant craniofacial involvement. J Oral Maxillofac Surg. 2012. 70: 102-18
11. Kos M, Luczak K, Godzinski J, Klempous J. Treatment of monostotic fibrous dysplasia with pamidronate. J Craniomaxillofac Surg. 2004. 32: 10-5
12. Kruse A, Pieles U, Riener MO, Zunker C, Bredell MG, Grätz KW. Craniomaxillofacial fibrous dysplasia: A 10 year database 1996-2006. Br J Oral Maxillofac Surg. 2009. 47: 302-5
13. Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012. 7: S2
14. Lei P, Bai H, Yu W, Liu Q. Surgical treatment of skull fibrous dysplasia. Surg Neurol. 2009. 72: 17S-20
15. Panda NK, Parida PK, Sharma R, Jain A, Bapuraj JR. A clinicoradiologic analysis of symptomatic craniofacial fibroosseous lesions. Otolaryngol Head Neck Surg. 2007. 136: 928-33
16. Park BY, Cheon YW, Kim YO, Pae NS, Lee WJ. Prognosis for craniofacial fibrous dysplasia after incomplete resection: AGE and serum alkaline phosphatase. Int J Oral Maxillofac Surg. 2010. 39: 221-6
17. Penn DL, Tartarini RJ, Glass CH, De Girolami U, Zamani AA, Dunn IF. Natural history of cranial fibrous dysplasia revealed during long-term follow-up: Case report and literature review. Surg Neurol Int. 2017. 8: 209
18. Rahman AM, Madge SN, Billing K, Anderson PJ, Leibovitch I, Selva D. Craniofacial fibrous dysplasia: Clinical characteristics and long-term outcomes. Eye (Lond). 2009. 23: 21
19. Reed RJ. Fibrous dysplasia of bone. A review of 25 cases. Arch Pathol. 1963. 75: 480-95
20. Schlumberger HG. Fibrous dysplasia of single bones (monostotic fibrous dysplasia). Mil Surg. 1946. 99: 504-27
21. Valentini V, Cassoni A, Terenzi V, Monaca MD, Fadda MT, Zadeh OR. Our experience in the surgical management of craniofacial fibrous dysplasia: What has changed in the last 10 years?. Acta Otorhinolaryngol Ital. 2017. 37: 436-4
22. Winn HR.editors. Youmans Neurological Surgery. Netherlands: Elsevier; 2011. p.
23. Zanotti B, Zingaretti N, Verlicchi A, Robiony M, Alfieri A, Parodi PC. Cranioplasty: Review of materials. J Craniofac Surg. 2016. 27: 2061-72

