

- Department of Neurosurgery, Narayana Health,
- Department of Endocrinology, Mazumdar Shaw Medical Center, Narayana Health, Bengaluru, Karnataka, India.
Correspondence Address:
Subramanian Kannan, Department of Endocrinology, Mazumdar Shaw Medical Center, Narayana Health, Bengaluru, Karnataka, India.
DOI:10.25259/SNI_681_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Mrudul Mohinish Bhatjiwale1, Komal Prasad Chandrachari1, Subramanian Kannan2. Dorsal vertebral body tumor and non-compressive quadriparesis – A rare case report of a phosphaturic mesenchymal tumor. 30-Sep-2022;13:452
How to cite this URL: Mrudul Mohinish Bhatjiwale1, Komal Prasad Chandrachari1, Subramanian Kannan2. Dorsal vertebral body tumor and non-compressive quadriparesis – A rare case report of a phosphaturic mesenchymal tumor. 30-Sep-2022;13:452. Available from: https://surgicalneurologyint.com/surgicalint-articles/11898/
Date of Submission
28-Jul-2022
Date of Acceptance
14-Sep-2022
Date of Web Publication
30-Sep-2022
Abstract
Background: Phosphaturic mesenchymal tumors cause renal phosphate wasting leading to hypophosphatemia manifesting as bone pain and fractures. About 95% of these tumors involve the extremities and the appendicular skeleton, with spinal tumors being exceedingly rare. We describe a case of non-compressive quadriparesis, caused by a thoracic vertebral body phosphaturic mesenchymal tumor (PMT).
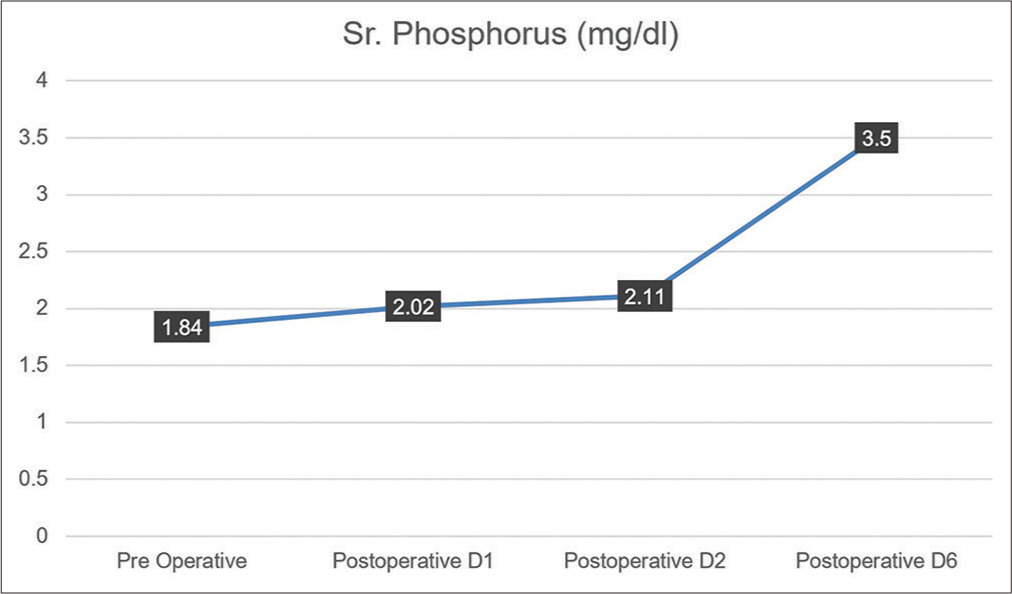
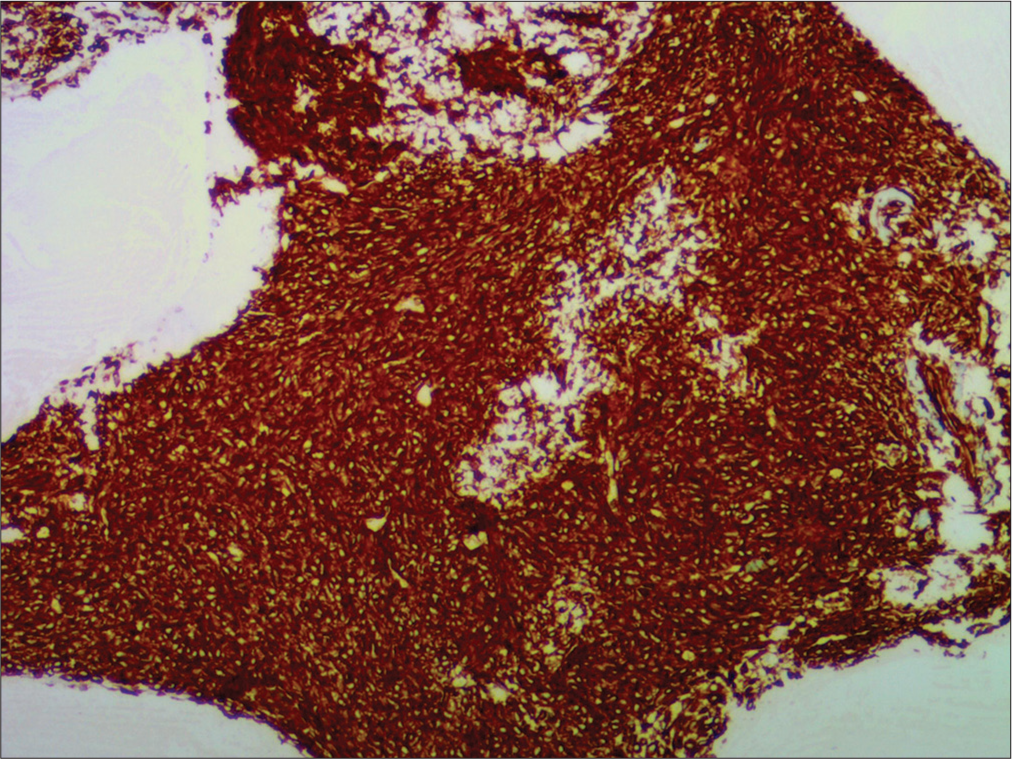
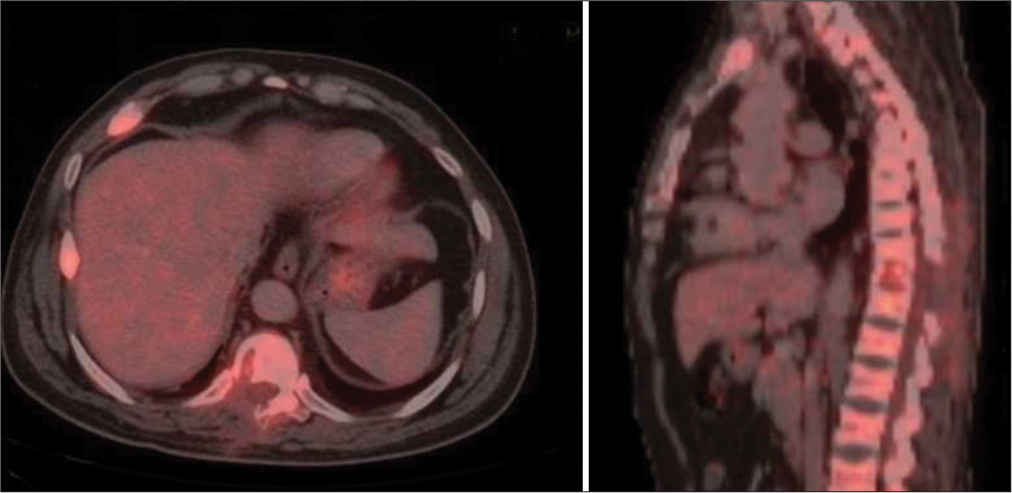
Case Description: A 34-year-old gentleman presented with a 3-year history of gradually progressive quadriparesis, predominantly involving proximal muscles. Magnetic resonance imaging neither showed evidence of compressive lesions nor myelopathy. On routine biochemistry, he was found to have hypophosphatemia and phosphaturia, with serum phosphorus levels of 1.84 mg/dl, and fractional excretion of phosphorus >5%. A DOTATOC positron emission tomography scan suggested the possibility of a PMT in the D10 vertebral body with a corresponding hyperdense/sclerotic focus on non-contrast computed tomography. After instituting phosphate replacement therapy and complete surgical excision of the hyperdense focus, he made a full neurological recovery. His phosphate levels normalized without the need for supplements. Histopathological examination showed spindle cells with positive staining for Vimentin, a mesenchymal cell marker. At 5 years of follow-up, the patient continues to be asymptomatic with a full return to normal function and no residual weakness.
Conclusion: PMTs involving the spine are a rare reversible cause of non-compressive quadriparesis. Early diagnosis and excision of the lesion correct the hypophosphatemia and result in complete neurological recovery.
Keywords: Hypophosphatemia, Mesenchymal, Phosphaturic, Quadriparesis, Tumor, Vertebral
INTRODUCTION
Quadriparesis is a symptom of varying etiology and is investigated, diagnosed, and treated by the neurologist, the neurosurgeon, the spine surgeon, and the physician, alike. Proximal muscle weakness narrows down the spectrum which ranges from infectious agents including influenza, Epstein-Barr virus, and Human immunodeficiency virus in patients with associated risk factors to the use of alcohol or steroids. Neurologic conditions that can cause proximal weakness include cerebrovascular disease, brain as well as spinal tumors, myopathies, demyelinating disorders (i.e., multiple sclerosis, Guillain-Barré syndrome), and neuromuscular disorders (i.e., myasthenia gravis, botulism).[
A phosphaturic mesenchymal tumor (PMT) is a neoplasm most commonly of mesenchymal origin, arising from and involving bone or soft tissue. The tumor produces a phosphaturic substance, fibroblast growth factor-23 (FGF23) that causes renal phosphate loss and subsequent systemic phosphate depletion, commonly leading to oncogenic osteomalacia, which is its most common presenting feature. Rarely hypophosphatemia may present as proximal muscle weakness and must be considered as a differential for the same. The diagnosis is often delayed due to the lack of clinical suspicion and nonspecific nature of the symptoms and, hence, it is important to be aware of this entity as one of the easily reversible causes of hypophosphatemia and resultant quadriparesis.
Surgery for spinal tumors is usually performed to decompress the neural elements and/or for spinal stabilization. We present a rare case where a hemivertebrectomy was performed to excise a small vertebral body PMT that was the cause of a paraneoplastic syndrome resulting in non-compressive quadriparesis.
CASE PRESENTATION
A 34-year-old gentleman presented to the clinic with complaints of weakness of all limbs, proximal greater than distal, for the past 3 years along with nonspecific mild back pain. His weakness had been gradually progressive, and for the past 1 year, he could no longer walk independently and needed support for daily activities. For the past 10 months, he was bedridden and severely functionally restricted. Examination revealed findings corroborated with the history. While the tone was normal, muscle power in the shoulders and hips bilaterally was ⅗ (MRC Grading), bilateral elbows and knees ⅘ and bilateral ankles, wrists, and grip 5/5 suggesting a predominant proximal muscle weakness. Sensations over the body and deep tendon reflexes were normal. An magnetic resonance imaging (MRI) whole spine was performed that revealed a normal result with no structural pathology that could explain the weakness.
Laboratory tests revealed hypophosphatemia (serum phosphorus levels of 1.84 mg/dl), which explained his proximal muscle weakness, with >5% fractional phosphorus excretion in the urine. An avid focus on DOTATOC positron emission tomography (PET) scan [
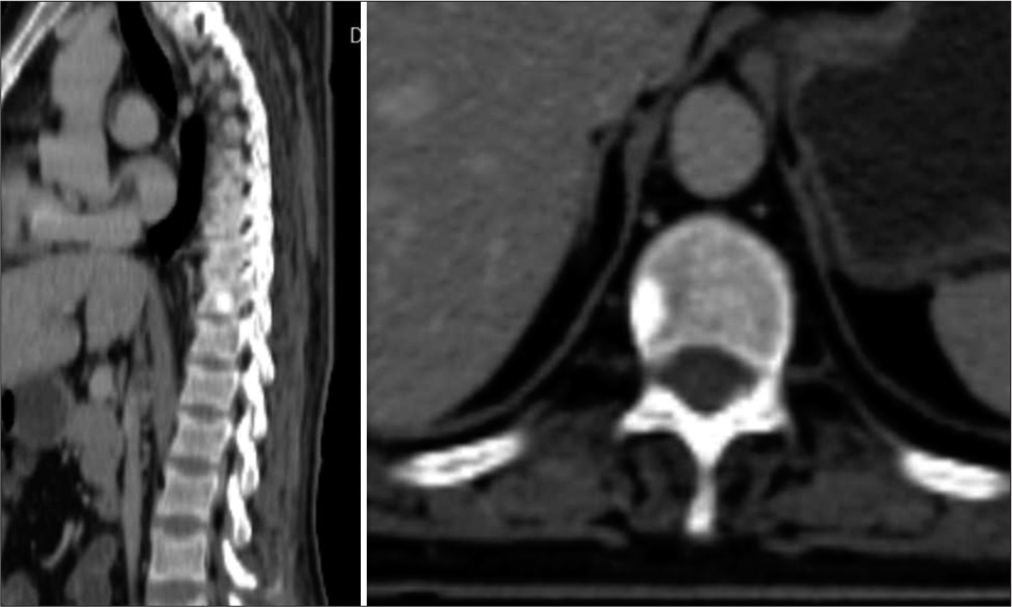
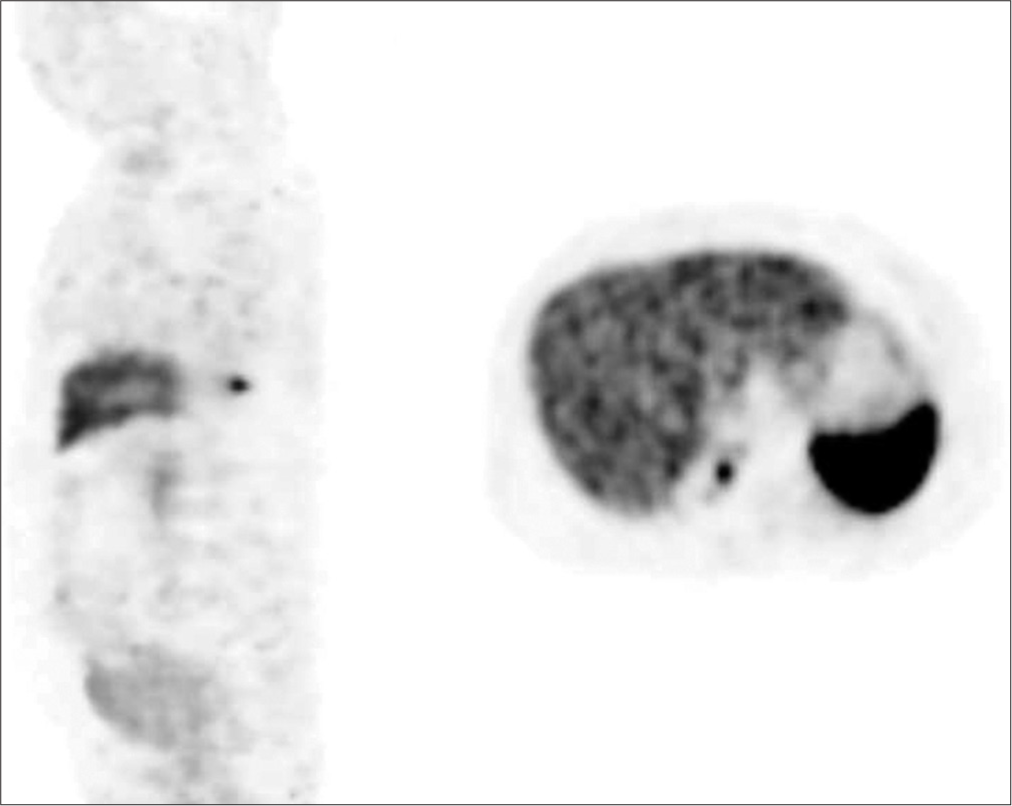
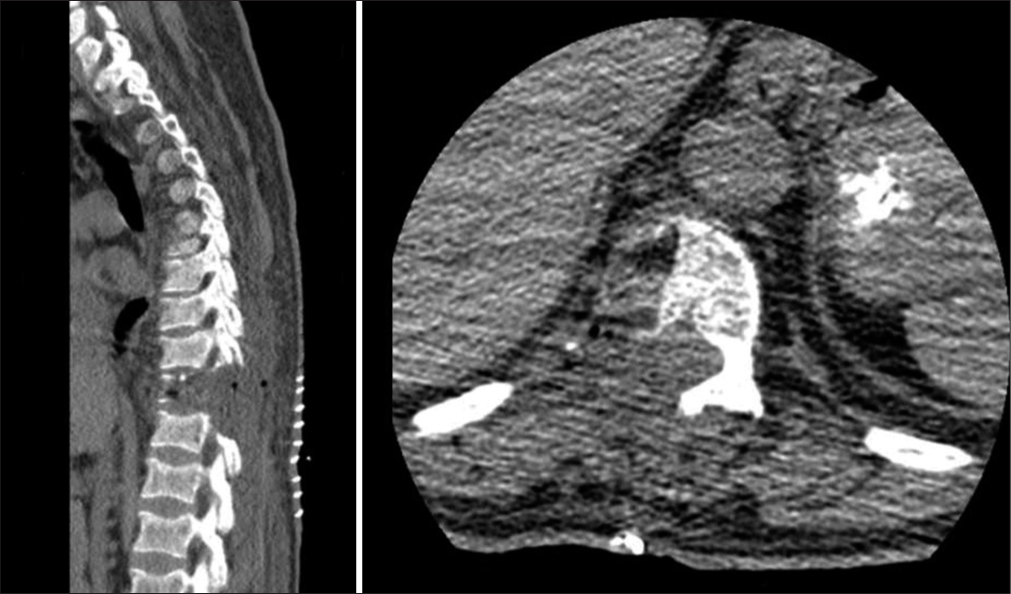
DISCUSSION
PMTs are rare tumors of mesenchymal origin. Most tumors are found in soft tissues, with bone being the second most common site.[
Presenting features
They are termed “phosphaturic” as a factor produced by the tumor cells, FGF23 also termed “phosphatonin,” promotes renal phosphate excretion, leading to chronic and rarely acute hypophosphatemia, in turn, leading to generalized weakness, malaise, and in severe cases hypotension, congestive heart failure, rhabdomyolysis, respiratory failure, seizures, altered mental status, and even death.[
Due to the rarity of this condition, most cases, as in our case, usually suffer for years before being diagnosed. Most are treated nonspecifically and symptomatically for long periods at primary and secondary care centers, before finally being diagnosed.
Pathogenesis and the role of FGF23
A high FGF23 level in a patient with normal renal function, and hypophosphatemia, with or without osteomalacia, is suggestive of a PMT.
FGF23 was first discovered in 2000, in autosomal dominant hypophosphatemic rickets by White et al.[
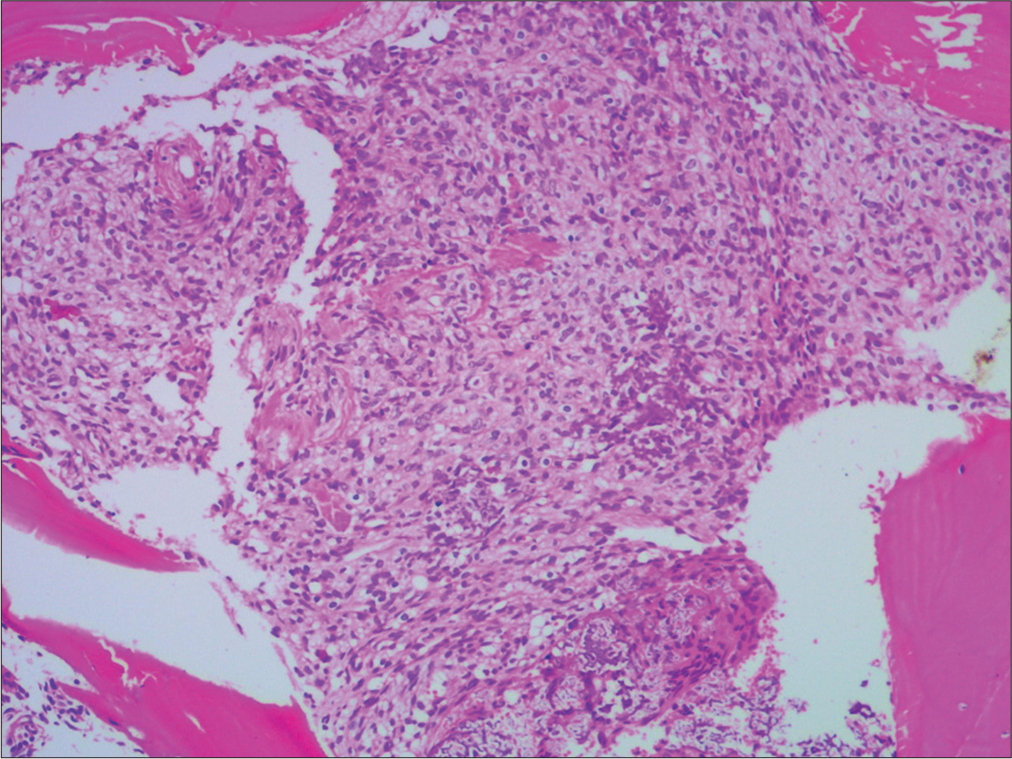
Histopathology
Histologically, PMTs are characterized by a proliferation of bland spindle cells associated with a variable amount of a “smudgy” calcified matrix. Mitosis and atypia are extremely rare and most tumors are benign. However, a very small percentage does exhibit nuclear atypia and a high Ki-67 index, and this subset might also be clinically malignant. There is also a small percentage of tumors that may be non-phosphaturic, and these pose a diagnostic challenge. Agaimy et al. in their series showed consistent expression of CD56 (100%), ERG (90%), and SATB2 (90%) on immunohistochemistry of PMTs.[
Management
In phosphaturic or “classical” PMTs, before surgery or when surgery is not indicated, oral phosphate and calcitriol can alleviate symptoms and metabolic imbalance.[
Complete tumor resection confers a good prognosis in most patients. However, surveillance for recurrence and metastasis is necessary, although most tumors do not recur or metastasize.
CONCLUSION
PMTs must be included in the differential diagnosis of patients presenting with quadriparesis with low serum phosphate levels. Elevated serum FGF-23 levels are diagnostic with a fair level of accuracy. Locating the tumor might prove challenging due to its small size at presentation and the possibility of involving almost any mesenchyme-derived structure in the body. DOTATOC PET CT can help locate these tumors when clinical and laboratory findings strongly suggest the same. Once diagnosed, complete surgical excision is the recommended treatment. Phosphate supplementation can hasten symptomatic recovery in operated cases and might alleviate the symptoms to a certain extent in inoperable or high-risk cases. The return of phosphate levels to normal usually provides excellent results and patients returning with recurrence are not common.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Agaimy A, Michal M, Chiosea S, Petersson F, Hadravsky L, Kristiansen G. Phosphaturic mesenchymal tumors: Clinicopathologic, immunohistochemical and molecular analysis of 22 cases expanding their morphologic and immunophenotypic spectrum. Am J Surg Pathol. 2017. 41: 1371-80
2. Ghorbani-Aghbolaghi A, Darrow MA, Wang T. Phosphaturic mesenchymal tumor (PMT): Exceptionally rare disease, yet crucial not to miss. Autops Case Rep. 2017. 7: 32-7
3. Jüppner H. Phosphate and FGF-23. Kidney Int Suppl. 2011. 79: S24-7
4. Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005. 71: 1327-36
5. White KE, Evans WE, O’Riordan JL, Speer MC, Econs MJ, Lorenz-Depiereux B. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000. 26: 345-8
6. Zuo QY, Wang H, Li W, Niu XH, Huang YH, Chen J. Treatment and outcomes of tumor-induced osteomalacia associated with phosphaturic mesenchymal tumors: Retrospective review of 12 patients. BMC Musculoskelet Disord. 2017. 18: 403

