

- Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, TX 77030, USA
Correspondence Address:
Sandi Lam
Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, TX 77030, USA
DOI:10.4103/2152-7806.166761
Copyright: © 2015 Lam S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.How to cite this article: Lam S, Reddy GD, Mayer R, Lin Y, Jea A. Eosinophilic granuloma/Langerhans cell histiocytosis: Pediatric neurosurgery update. Surg Neurol Int 07-Oct-2015;6:
How to cite this URL: Lam S, Reddy GD, Mayer R, Lin Y, Jea A. Eosinophilic granuloma/Langerhans cell histiocytosis: Pediatric neurosurgery update. Surg Neurol Int 07-Oct-2015;6:. Available from: http://surgicalneurologyint.com/surgicalint_articles/eosinophilic-granulomalangerhans-cell-histiocytosis-pediatric/
ILLUSTRATIVE CASES
Case 1
A 23-month-old female was admitted to the neurosurgery service with a 3-month history of a progressively enlarging neck mass. There was associated redness, swelling, and tenderness to palpation, but no neurological deficits on examination. A noncontrast computed tomography (CT) scan of the neck and magnetic resonance imaging (MRI) with contrast showed an osteolytic contrast-enhancing lesion primarily involving the C2 posterior elements, with a compressive circumferential epidural component extending from C2 to C5 [
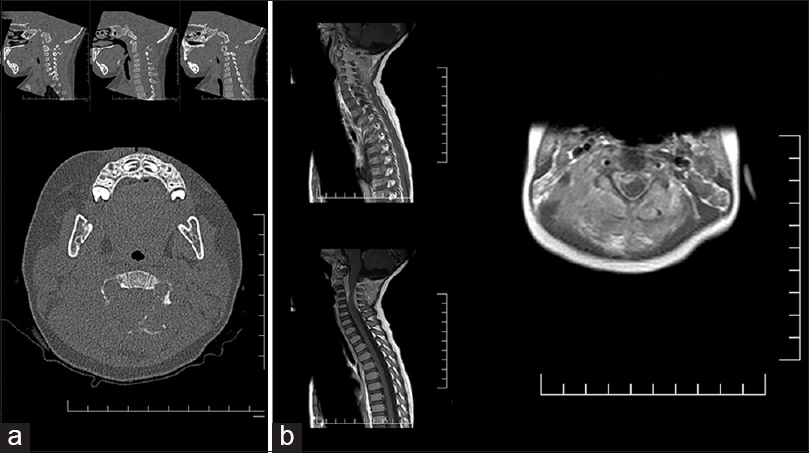
Case 2
A 17-year-old male was admitted to the neurosurgery service with a 6-week history of a progressively enlarging scalp mass. There were tenderness and intermittent bleeding from the ulcerated lesion, with no focal neurologic deficits on examination. A noncontrast CT scan of the head and MRI brain, with and without contrast, showed a large transosseous contrast-enhancing lesion of the right frontal bone [
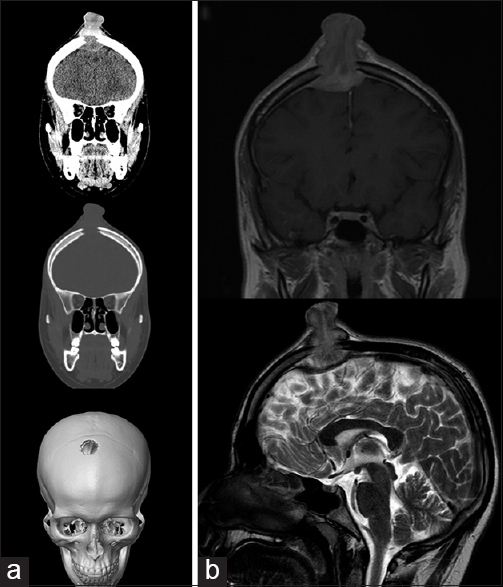
Figure 2
Images for the patient in case 2. (a) Computed tomography head without contrast. Coronal brain window (top), coronal bone window (middle), and three-dimensional skull reconstruction (bottom) images. (b) Magnetic resonance imaging brain. Coronal postcontrast T1-weighted image (top) and sagittal T2-weighted (bottom) images
REVIEW
Overview and nosology of Langerhans cell histiocytosis
LCH is a rare heterogeneous illness characterized by the proliferation of dendritic cells with LCH morphology. LCH refers to a spectrum of diseases, from a localized lesion to a diffuse multiorgan pathology.[
Etiology and pathophysiology
As mentioned above, the proliferation of a myeloid-derived precursor dendritic cell with the characteristics of a LC is what characterizes LCH.[
The central debate on the pathogenesis of LCH is whether it is a reactive immune response or a neoplasm. Support for the neoplastic theory includes the monoclonality of the pathologic cells (though this also can be seen with reactive immune processes),[
Epidemiology
EG that affects the bone is the most common subtype of LCH, representing an estimated 60–80% of cases. EG can be single or multifocal; it most commonly affects the calvarium, but can also present in the vertebrae, ribs, long bones, and mandible.[
Clinical presentation, workup, and diagnosis
Radiography demonstrates a sharply demarcated osteolytic lesion of the underlying bone.[
The differential for an osteolytic mass is broad and includes neoplastic processes such as metastatic lesions, primary bone tumors, including osteosarcoma or Ewing's sarcoma, neuroblastoma, rhabdomyosarcoma, lymphoma and primitive neuroectodermal tumors. It also includes infections such as osteomyelitis or abscesses, fibrous dysplasia, and cystic lesions such as aneurysmal bone cysts or dermoid cysts. Vascular processes, such as venous lakes, hemangiomas, and angiomatosis, are also in the differential, as well as developmental anomalies such as neurenteric cysts and encephaloceles. Tissue biopsy is necessary for definitive diagnosis. The classic radiographic presentation of vertebra plana (not shown in our more dramatic Case 1) is not considered pathognomonic of LCH, as complete vertebral collapse can be seen with other diagnoses such as Ewing's sarcoma and infection.
Histopathologically, LCH lesions show a proliferation of LC-type cells in a milieu of lymphocytes, macrophages, and eosinophils. In 1987, the Histiocyte Society established the diagnostic criteria for a diagnosis of LCH, which required the identification of CD1a on immunohistochemistry or Birbeck granules on electron microscopy.[
Workup should include a systemic survey to identify any other potential sites of involvement, as management recommendations and prognosis vary depending on the number and type of organs involved in LCH.[
Treatment
LCH of the spine is rare. Treatment patterns in the literature include observation, complete surgical excision with fixation, and radiotherapy. Bertram et al. reviewed the literature of spine EG (n = 53) and found that most cases resolved without treatment. Immobilization and observation are recommended in cases without spinal instability or neurological deficit.[
Solitary EG of the calvarial vault without invasion into neurological structures has a favorable prognosis. These lesions typically are managed with surgical curettage or excision.[
The Histiocyte Society recommends more aggressive treatment in cases of multifocal bone disease or disease that involves “CNS-risk” sites (odontoid, vertebrae with intraspinal soft-tissue extension, facial bones, skull base, orbit, oral cavity).[
Prognosis
In a large study from South Korea (n = 603), 5-year overall survival was 99.8% in those with single-system LCH, 98.4% for multisystem LCH without risk organ involvement, and 77% for multisystem LCH with risk organ involvement.[
CONCLUSION
LCH describes a heterogeneous mixture of pathologies and should be on the differential for any osteolytic soft-tissue mass. Definitive diagnosis requires tissue biopsy, and treatment options vary from observation to resection and chemotherapy. Prognosis is good, and recurrence rates are low, particularly for patients with single bone lesions.
References
1. Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev. 2010. 36: 354-9
2. Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared to epidermal Langerhans cells. J Immunol. 2010. 184: 4557-67
3. Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis: Pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005. 35: 103-15
4. Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010. 116: 1919-23
5. Bank MI, Rengtved P, Carstensen H, Petersen BL. Langerhans cell histiocytosis: An evaluation of histopathological parameters, demonstration of proliferation by Ki-67 and mitotic bodies. APMIS. 2003. 111: 300-8
6. Bertram C, Madert J, Eggers C. Eosinophilic granuloma of the cervical spine. Spine (Phila Pa 1976). 2002. 27: 1408-13
7. Binning MJ, Brockmeyer DL. Novel multidisciplinary approach for treatment of Langerhans cell histiocytosis of the skull base. Skull Base. 2008. 18: 53-8
8. Cantu MA, Lupo PJ, Bilgi M, Hicks MJ, Allen CE, McClain KL. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS One. 2012. 7: e43257-
9. da Costa CE, Szuhai K, van Eijk R, Hoogeboom M, Sciot R, Mertens F. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer. 2009. 48: 239-49
10. D’Ambrosio N, Soohoo S, Warshall C, Johnson A, Karimi S. Craniofacial and intracranial manifestations of Langerhans cell histiocytosis: Report of findings in 100 patients. AJR Am J Roentgenol. 2008. 191: 589-97
11. De Angulo G, Nair S, Lee V, Khatib Z, Ragheb J, Sandberg DI. Nonoperative management of solitary eosinophilic granulomas of the calvaria. J Neurosurg Pediatr. 2013. 12: 1-5
12. Denaro L, Longo UG, Papalia R, Di Martino A, Maffulli N, Denaro V. Eosinophilic granuloma of the pediatric cervical spine. Spine (Phila Pa 1976). 2008. 33: E936-41
13. Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: Abundant expression of cytokines relevant to disease and treatment. Blood. 1999. 94: 4195-201
14. Egeler RM, Thompson RC, Voûte PA, Nesbit ME. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop. 1992. 12: 811-4
15. Erly WK, Carmody RF, Dryden RM. Orbital histiocytosis X. AJNR Am J Neuroradiol. 1995. 16: 1258-61
16. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol. 2001. 23: 54-6
17. Gadner H, Minkov M, Grois N, Pötschger U, Thiem E, Aricò M. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013. 121: 5006-14
18. Geissmann F, Lepelletier Y, Fraitag S, Valladeau J, Bodemer C, Debré M. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood. 2001. 97: 1241-8
19. Geissmann F. Histiocytosis and the Mononuclear Phagocyte System. Proceedings of the 24th Annual Meeting of the Histiocyte Society, October 1-3, Berlin, Germany. 2008. p.
20. Greenlee JD, Fenoy AJ, Donovan KA, Menezes AH. Eosinophilic granuloma in the pediatric spine. Pediatr Neurosurg. 2007. 43: 285-92
21. Gunny R, Clifton A, Al-Memar A. Spontaneous regression of supratentorial intracerebral Langerhans’ cell histiocytosis. Br J Radiol. 2004. 77: 685-7
22. Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013. 121: 1495-500
23. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013. 60: 175-84
24. . Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet. 1987. 1: 208-9
25. Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: Diagnosis, natural history, management, and outcome. Cancer. 1999. 85: 2278-90
26. Jeziorski E, Senechal B, Molina TJ, Devez F, Leruez-Ville M, Morand P. Herpes-virus infection in patients with Langerhans cell histiocytosis: A case-controlled sero-epidemiological study, and in situ analysis. PLoS One. 2008. 3: e3262-
27. Jubran RF, Marachelian A, Dorey F, Malogolowkin M. Predictors of outcome in children with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005. 45: 37-42
28. Karagoz Guzey F, Bas NS, Emel E, Alatas I, Kebudi R. Polyostotic monosystemic calvarial and spinal Langerhans’ cell histiocytosis treated by surgery and chemotherapy. Pediatr Neurosurg. 2003. 38: 206-11
29. Kasper EM, Aguirre-Padilla DH, Alter RY, Anderson M. Histiocytosis X: Characteristics, behavior, and treatments as illustrated in a case series. Surg Neurol Int. 2011. 2: 57-
30. Kellenberger CJ, Epelman M, Miller SF, Babyn PS. Fast STIR whole-body MR imaging in children. Radiographics. 2004. 24: 1317-30
31. Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK. Langerhans’ cell histiocytosis (histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatric and adult cases. Cancer. 1995. 76: 2471-84
32. Kim BE, Koh KN, Suh JK, Im HJ, Song JS, Lee JW. Clinical features and treatment outcomes of Langerhans cell histiocytosis: A nationwide survey from Korea histiocytosis working party. J Pediatr Hematol Oncol. 2014. 36: 125-33
33. Lee HJ, Ahn BC, Lee SW, Lee J. The usefulness of F-18 fluorodeoxyglucose positron emission tomography/computed tomography in patients with Langerhans cell histiocytosis. Ann Nucl Med. 2012. 26: 730-7
34. Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, Letterer-Siwe disease, and Schüller-Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol. 1953. 56: 84-102
35. Mc Dermott R, Ziylan U, Spehner D, Bausinger H, Lipsker D, Mommaas M. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Mol Biol Cell. 2002. 13: 317-35
36. McClain K, Jin H, Gresik V, Favara B. Langerhans cell histiocytosis: Lack of a viral etiology. Am J Hematol. 1994. 47: 16-20
37. Meyer JS, De Camargo B. The role of radiology in the diagnosis and follow-up of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998. 12: 307-26
38. Minkov MGrois NMcClain KNanduri VRodriguez-Galindo CSimonitsch-Klupp ILast accessed on 2015 Apr 01. Available from: http://www.histiocytesociety.org/document.doc?id=290.
39. Morimoto A, Ikushima S, Kinugawa N, Ishii E, Kohdera U, Sako M. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. 2006. 107: 613-9
40. Mueller WP, Melzer HI, Schmid I, Coppenrath E, Bartenstein P, Pfluger T. The diagnostic value of 18F-FDG PET and MRI in paediatric histiocytosis. Eur J Nucl Med Mol Imaging. 2013. 40: 356-63
41. Munn SE, Olliver L, Broadbent V, Pritchard J. Use of indomethacin in Langerhans cell histiocytosis. Med Pediatr Oncol. 1999. 32: 247-9
42. Oliveira M, Steinbok P, Wu J, Heran N, Cochrane D. Spontaneous resolution of calvarial eosinophilic granuloma in children. Pediatr Neurosurg. 2003. 38: 247-52
43. Park SH, Park J, Hwang JH, Hwang SK, Hamm IS, Park YM. Eosinophilic granuloma of the skull: A retrospective analysis. Pediatr Neurosurg. 2007. 43: 97-101
44. Phillips M, Allen C, Gerson P, McClain K. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2009. 52: 97-101
45. Postini AM, Andreacchio A, Boffano M, Pagano M, Brach Del Prever A, Fagioli F. Langerhans cell histiocytosis of bone in children: A long-term retrospective study. J Pediatr Orthop B. 2012. 21: 457-62
46. Raab P, Hohmann F, Kühl J, Krauspe R. Vertebral remodeling in eosinophilic granuloma of the spine. A long-term follow-up. Spine (Phila Pa 1976). 1998. 23: 1351-4
47. Schouten B, Egeler RM, Leenen PJ, Taminiau AH, van den Broek LJ, Hogendoorn PC. Expression of cell cycle-related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2002. 24: 727-32
48. Senechal B, Elain G, Jeziorski E, Grondin V, Patey-Mariaud de Serre N, Jaubert F. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007. 4: e253-
49. Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH. Langerhans’-cell histiocytosis (histiocytosis X) – A clonal proliferative disease. N Engl J Med. 1994. 331: 154-60
50. Yeom JS, Lee CK, Shin HY, Lee CS, Han CS, Chang H. Langerhans’ cell histiocytosis of the spine. Analysis of twenty-three cases. Spine (Phila Pa 1976). 1999. 24: 1740-9

