

- Department of Neurosurgery, University Hospital Center Zagreb, School of Medicine, Zagreb, Croatia
- Department of Pathology, University Hospital Center Zagreb, School of Medicine, Zagreb, Croatia
- Department of Neurosurgery, Dubrovnik General Hospital, Dubrovnik, Croatia
Correspondence Address:
Jakob Nemir
Department of Neurosurgery, University Hospital Center Zagreb, School of Medicine, Zagreb, Croatia
DOI:10.4103/2152-7806.196933
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Jakob Nemir, Ines Trninic, Kresimir S. Duric, Antonia Jakovcevic, Goran Mrak, Josip Paladino. Extranodal right-optic nerve Rosai–Dorfman disease: A rare localization case report. 28-Dec-2016;7:
How to cite this URL: Jakob Nemir, Ines Trninic, Kresimir S. Duric, Antonia Jakovcevic, Goran Mrak, Josip Paladino. Extranodal right-optic nerve Rosai–Dorfman disease: A rare localization case report. 28-Dec-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/extranodal-right%e2%80%91optic-nerve-rosai-dorfman-disease-a-rare-localization-case-report/
Date of Submission
09-Feb-2016
Date of Acceptance
17-Jun-2016
Date of Web Publication
28-Dec-2016
Abstract
Background:Rosai–Dorfman is a rare disease that usually occurs in young adults. It is characterized with massive painless cervical lymphadenopathy and histiocyte proliferation. Isolated intracranial involvement is extremely rare. Our aim is to present a new rare case of extranodal Rosai–Dorfman disease that involved the right optic nerve in a 4-year-old boy.
Case Description:A 4-year-old boy with right-sided convergent strabismus and amblyopia lasting for 1 year was treated at the Department of pediatric ophthalmology. Initial optical fundus examination was normal. Examination repeated after 1 year noted the atrophy of the optic nerve papilla. Visual evoked potentials of the right eye showed normal findings of prechiasmatic visual pathway with severe dysfunction of the right optic nerve. Magnetic resonance imaging (MRI) of the brain and orbits showed expansive changed and elongated right optic nerve with contrast enhancement, and smaller lesion in the right temporal operculum region visible in T2 and fluid-attenuated inversion recovery sequence. Through small eyebrow “keyhole” osteoplastic frontoorbital craniotomy the fusiform enlarged (to 2 cm) right optic nerve was identified, resected between the eyeball and optic chiasm, and transferred for pathohistological analysis. Early postoperative course had no complications. Histological, immunohistochemical, and ultrastructural analyses revealed extranodal Rosai–Dorfman disease. Right periorbital edema was verified on the 7th postoperative day and regressed to supportive therapy. Control multi slice computed tomography (MSCT) and MRI of endocranium and orbits showed total tumor removal with no signs of complications.
Conclusion:Although rare, extranodular intracranial Rosai–Dorfman disease should be taken into account in the differential diagnosis of intracranial and intraorbital lesions, especially in the pediatric age group.
Keywords: Extranodal, optic nerve, pediatric tumor, Rosai–Dorfman disease
INTRODUCTION
Rosai–Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy (SHML) is an uncommon benign histiocytic proliferative disorder of unknown origin.[
CASE DESCRIPTION
History and examination
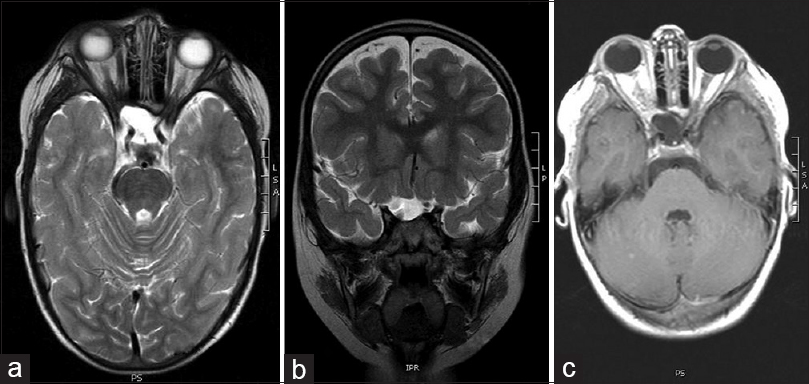
A 4-year-old boy presented with right-sided convergent strabismus and amblyopia that lasted for 1 year and was admitted to the Department of Pediatric Ophthalmology. His medical history was unremarkable except for the thrombocytopenia treated at 2 years of age. Before admission, he had regular ophthalmological exams and was treated with corrective glasses. An eye examination was performed by an ophthalmologist. Visual acuity was normal on his left eye, but visual acuity on his right eye was poor, he did not have the sense of light. Both eye bulb motility was normal with good pupil function. Initial optical fundus examination was normal. The cover test was positive on his right eye with right-sided convergent strabismus. Visual field was not done because of the patient's age. On the last ophthalmologic control exam, due to the right-sided atrophy of the optic nerve papilla and amblyopia, performing visual evoked potentials (VEP) and magnetic resonance imaging (MRI) were recommended. VEP of the right eye showed normal findings of prechiasmatic visual pathway with severe dysfunction of the right optic nerve. MRI of the brain and orbits showed expansive changed and elongated right optic nerve with contrast enhancement; furthermore, a smaller lesion in the right temporal operculum region was visible in T2 and fluid-attenuated inversion recovery sequence, as well as a small oval lesion in the left cerebellar lobe [Figures
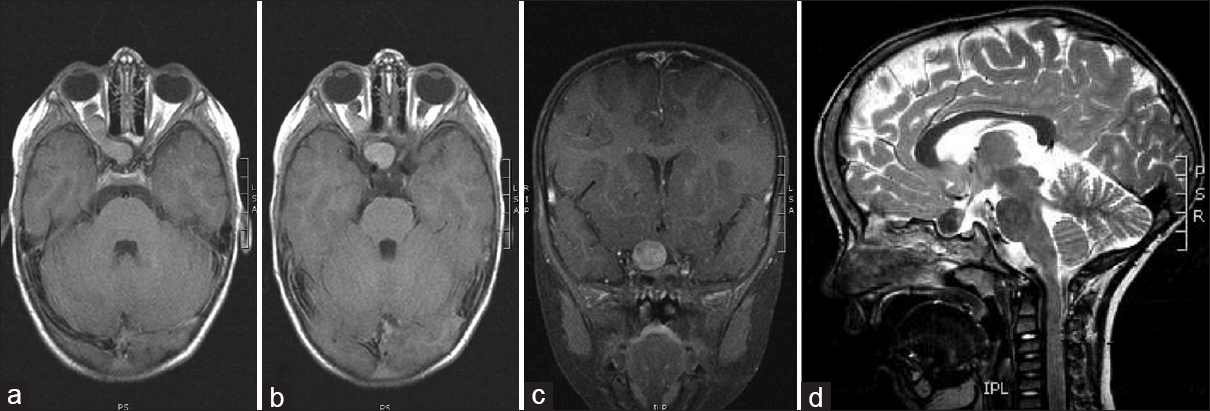
Surgical technique
The position of the patient was supine, with the head turned to the left at 15°, leaving the eyebrow as the most prominent point. Skin was incised through the eyebrow, medially up to the supraorbital notch leaving the supraorbital nerve intact. With one small burr hole at the superior temporal line, small supraorbital bone flap was performed which was 3 cm in width and 2.5 cm in height using a craniotome, including linear extensions over the supraorbital arch. Periorbita was detached from the bone and the bony flap was pushed down toward the orbit until the orbital roof brakes. With slight elevation of the bone, dura was detached from the orbital roof and the whole bone flap including supraorbital arch was removed in one piece. Using a diamond drill, the whole orbital roof was drilled out, optic canal widely opened, and anterior clinoid removed extraduraly. Periorbit was incised longitudinally, including annulus of Zinni and the dura over the optic nerve and frontobasaly, meticulously dissecting orbital muscles and nerves before reaching the optic nerve. Almost the whole optic nerve was thickened, and was irregularly shaped 2 mm from the eyeball and up to the chiasm. The nerve with the infiltrating tumor was cut leaving the normal white tissue at the resected planes of the nerve. Macroscopically, the tumor was firm, avascular, and had a gray-yellow color infiltrating the whole width of the optic nerve. There was no bleeding at the resected planes of the nerve and dura and periorbit was partly sutured and sealed. Bone flap was attached using microscrews, and the wound was closed in the standard manner.
Postoperative course
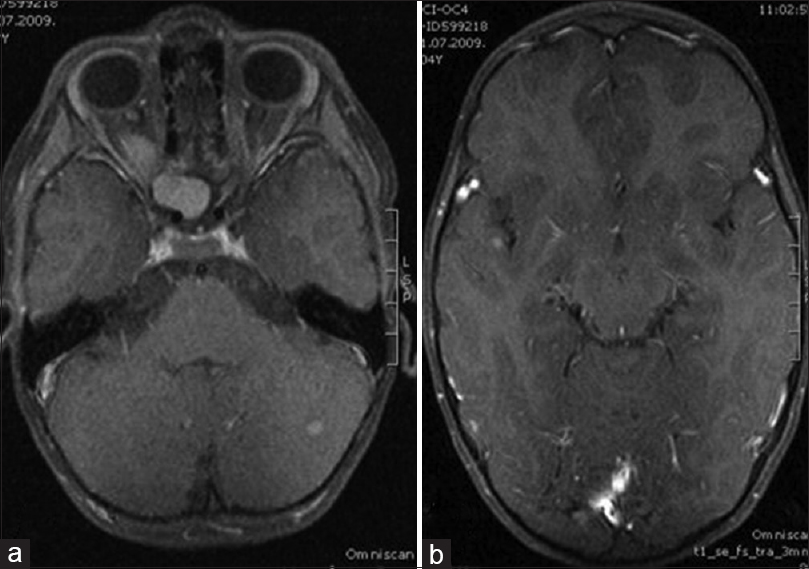
Early postoperative course was uneventful, except right-sided periorbital hematoma that spontaneously regressed few days later. Later, the patient received corneal ulcer refractory to treatment with topical antibiotic drops and cream for 3 months, after which the amniotic membrane transplant was performed to heal the ulcer completely. After 3 years of follow-up, MRI showed complete tumor removal without any sign of recurrence [
Histopathological workup
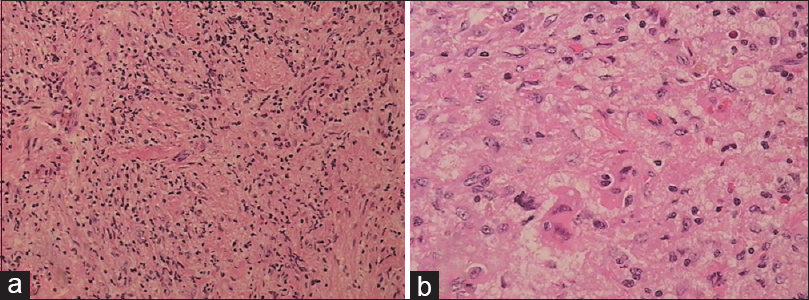
Microscopic examination of the surgical sample revealed a mainly histiocytic lesion in a fibrous background admixed with lesser populations of lymphocytes and plasma cells. The histiocytic cells showed evidence of emperipolesis. Immunohistochemically, tumor cells were CD68 and S100 positive and negative for langerin and CD1a. The phenotype was consistent with RDD [Figures
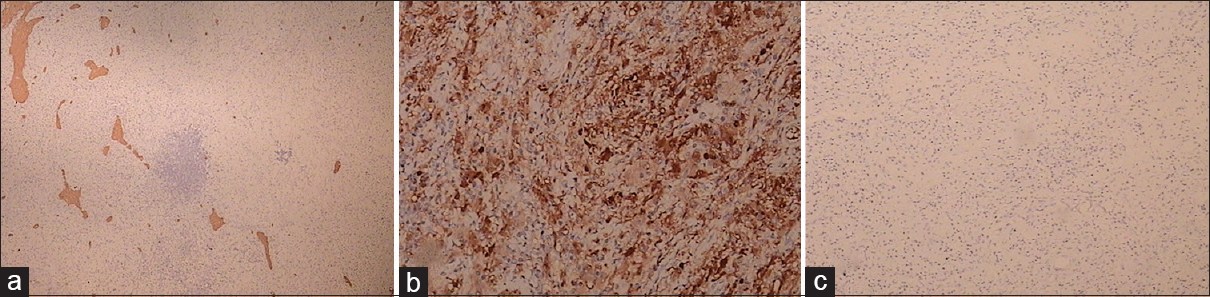
DISCUSSION
RDD is a rare histiocytic disorder initially described as a separate entity in 1969 by Rosai and Dorfman using the term SHML.[
CONCLUSION
Extranodular intracranial RDD should be taken into account in the differential diagnosis of intracranial and intraorbital lesions, especially in pediatric age group.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We thank Prof. Christopher D. M. Fletcher, MD, FRCPath, from the Department of pathology Brigham and Women's Hospital Boston, USA, for his support in diagnosis.
References
1. Al-Saad K, Thorner P, Ngan BY, Gerstle JT, Kulkarni AV, Babyn P. Extranodal Rosai-Dorfman disease with multifocal bone and epidural involvement causing recurrent spinal cord compression. Pediatr Dev Pathol. 2005. 8: 593-8
2. Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: A report of 11 cases. Mod Pathol. 2001. 14: 172-8
3. Carbone A, Passannante A, Gloghini A, Devaney KO, Rinaldo A, Ferlito A. Review of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) of head and neck. Ann Otol Rhinol Laryngol. 1999. 108: 1095-104
4. Carpenter RJ, Banks PM, Mc Donald TJ, Sanderson DR. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Report of a case with respiratory tract involvement. Laryngoscope. 1978. 88: 1963-9
5. Cooper SL, Chavis PS, Fortney JA, Watkins JM, Caplan MJ, Jenrette JM. A case of orbital Rosai-Dorfman disease responding to radiotherapy. J Pediatr Hematol Oncol. 2008. 30: 744-8
6. Deodhare SS, Ang LC, Bilbao JM. Isolated intracranial involvement in Rosai-Dorfman disease: A report of two cases and a review of the literature. Arch Pathol Lab Med. 1998. 122: 161-5
7. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990. 7: 19-73
8. Goodnight JW, Wang MB, Sercarz JA, Fu YS. Extranodal Rosai-Dorfman disease of the head and neck. Laryngoscope. 1996. 106: 253-6
9. Hargett C, Bassett T. Atypical presentation of sinus histiocytosis with massive lymphadenopathy as an epidural spinal cord tumor: A case presentation and literature review. J Spinal Disord Tech. 2005. 18: 193-6
10. Hazarika P, Nayak DR, Balakrishnan R, Kundaje HG, Rao PL. Rosai-Dorfman disease of the subglottis. J Laryngol Otol. 2000. 114: 970-3
11. Hollowell JP, Wolfla CE, Shah NC, Mark LP, Whittaker MH. Rosai-Dorfman disease causing cervical myelopathy. Spine. 2000. 25: 1453-6
12. Huang YC, Than HY, Jung SM, Chuang WY, Chuang CC, Hsu PW. Spinal epidural Rosai-Dorfman disease proceeded by relapsing uveitis: A case report with literature review. Spinal Cord. 2007. 45: 641-4
13. Juskevicius R, Finlay JL. Rosai-Dorfman disease of the parotid gland, cytologic and histopathologic findings with immunohistochemical correlation. Arch Pathol Lab Med. 2001. 125: 1348-50
14. Kidd DP, Revesz T, Miller NR. Rosai-Dorfman disease presenting with widespread intracranial and spinal cord involvement. Neurology. 2006. 67: 1551-5
15. Krafet SK, Honig M, Krishnamurthy S. Emperipolesis in the cerebrospinal fluid from a patient with Rosai-Dorfman disease. Diagn Cytopathol. 2007. 36: 67-8
16. Lauwers GY, Perez-Atayed A, Dorfman RF, Rosai . The digestive system manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy): Review of 11 cases. Hum Pathol. 2000. 31: 380-5
17. Petschner F, Walker UA, Scmitt-Graf A. Catastrophic systemic lupus erythematous with Rosai-Dorfman sinus histiocytosis. Successful treatment with anti-CD20/rituximab. Dtsch Med Wochenschr. 2001. 126: 998-1001
18. Pulsoni A, Anghel G, Falcucci P, Matera , R Pescarmona, Ribersani M. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Report of a case and literature Review. Am J Hematol. 2000. 69: 61-71
19. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: A newly recognized benign clinicopathological entity. Arch Pathol. 1969. 87: 63-70
20. Sanchez R, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: An analysis of 113 cases with special emphasis on its extranodal manifestations. Lab Invest. 1977. 36: 349-50
21. Scheel MM, Rady PL, Tyring SK, Pandya AG. Sinus histiocytosis with massive lymphadenopathy: Presentation as giant granuloma annulare and detection of human herpesvirus 6. J Am Acad Dermatol. 1997. 37: 643-6
22. Tian Y, Wang J, Li M, Lin S, Wang G, Wu Z, Ge M, Pirotte BJ. Rosai-Dorfman disease involving the central nervous system: Seven cases from one institute. Acta Neurochir. 2015. 157: 1565-71


