

- Departament of Surgical Specialities and Neurosurgery, Neurosurgery Teaching and Assistance Unit, Pedro Ernesto University Hospital, Rio de Janeiro, Brazil,
- Departament of Pathology, Pedro Ernesto University Hospital, Rio de Janeiro, Brazil.
DOI:10.25259/SNI_192_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Thainá Zanon Cruz1, Pedro Henrique Costa Ferreira-Pinto1, Ana Carolina Gonçalves Brito2, Leandro Ururahy1, Jefferson Trivino Sanchez1, Flavio Nigri1. Ganglioglioma of the cervicothoracic spinal cord in a patient with neurofibromatosis type 1: A case report. 28-Jun-2021;12:313
How to cite this URL: Thainá Zanon Cruz1, Pedro Henrique Costa Ferreira-Pinto1, Ana Carolina Gonçalves Brito2, Leandro Ururahy1, Jefferson Trivino Sanchez1, Flavio Nigri1. Ganglioglioma of the cervicothoracic spinal cord in a patient with neurofibromatosis type 1: A case report. 28-Jun-2021;12:313. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=10923
Date of Submission
24-Feb-2021
Date of Acceptance
26-May-2021
Date of Web Publication
28-Jun-2021
Abstract
Background: Gangliogliomas are rare tumors of the central nervous system. They are usually located intracranially and rarely in the spinal cord. There is no clear correlation between this tumor and neurofibromatosis type 1 (NF1) with only four cases described. The aim of this article is to describe one more case and add data to the literature regarding this rare association.
Case Description: An 8-year-old boy with NF1 presented progressive asymmetrical paraparesis (Grade 4 medical research council scale on the right leg and Grade 3 on the left leg). The cervicothoracic spinal magnetic resonance imaging demonstrated an intramedullary lesion from C4 to T4 vertebrae. The patient underwent a microsurgical resection. A partial resection was performed due to a drop in the motor evoked potential signal amplitude during dissection. Pathology report revealed a ganglioglioma (World Health Organization Grade I). Postoperatively, the patient evolved with worsening of the paraparesis. A few weeks later, he has improved his preoperative functional neurological state (better strength and gait). Adjuvant radiotherapy was not used. The patient is being followed up at the neurosurgery outpatient clinic.
Conclusion: This is another case of spinal ganglioglioma associated with NF1. The tumor must be included in the differential diagnosis of patients with NF1 and spinal lesions. Complete microsurgical resection remains the standard treatment for spinal gangliogliomas, however, in this specific case, it was decided to leave a portion of the tumor to prevent neurological damage. The prognosis and treatment of this condition associated with NF1 remains to be determined.
Keywords: Ganglioglioma, Microsurgery, Neurofibromatosis type 1, Spinal tumor
INTRODUCTION
Gangliogliomas are rare tumors of the central nervous system (CNS), corresponding to 2.7–3.8% of primary CNS tumors, composed of variable proportions of glial and neuronal elements.[
Neurofibromatosis type 1 (NF1) corresponds to an autosomal dominant genetic disorder in which mutations in the NF1 gene occur. The NF1 gene is located on the long arm of chromosome 17 (17q11.2).[
There are not enough studies in the literature to define whether the association of NF1 and gangliogliomas is incidental or not. After searching in the medical literature analysis and retrieval system online database, it was found four cases described. The first one was a T10-L2 spinal ganglioglioma in a 5-year-old female (Japan, 2011).[
CLINICAL PRESENTATION
An 8-year-old male patient was admitted to the pediatric unit complaining of weakness and pain of the lower limbs with occasional falls 1 year ago with significant worsening in the past 2 months. He had NF1 diagnosis since 2 years old with the presence of two of the seven National Institutes of Health criteria. The first criterion was more than six cafe-au-lait spots located on the lateral thoracic region with a diameter above 5 mm in a prepubertal individual. The second criterion was one plexiform neurofibroma in the lumbar region, confirmed after a biopsy performed in 2018. No other family members were diagnosed with NF1.
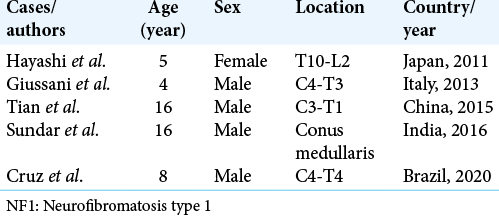
The physical examination revealed macrocrania, pectus excavatum, and leg length discrepancy. On neurological examination, he presented an asymmetrical paraparesis (Grade 4 medical research council [MRC] scale on the right leg and Grade 3 on the left leg) with spasticity, hyperreflexia, and bilateral Babinski signs. The patient was able to walk, but was unable to stay long due to weakness. The brain and spinal cord MRI were performed. The brain MRI revealed T2 hyperintense signal and T1 hypointense signal lesions, in the dorsal part of the right globus pallidus, cerebellar hemispheres, pons, and white matter. Contrast-enhanced T1-weighted images, with fat suppression technique of the cervicothoracic spinal MRI, demonstrated an intramedullary lesion from the superior endplate of the C4 vertebra to the inferior endplate of the T4 vertebra, with a peripheral isointense signal and an irregular hyperintense central suggesting an intramedullary mass [
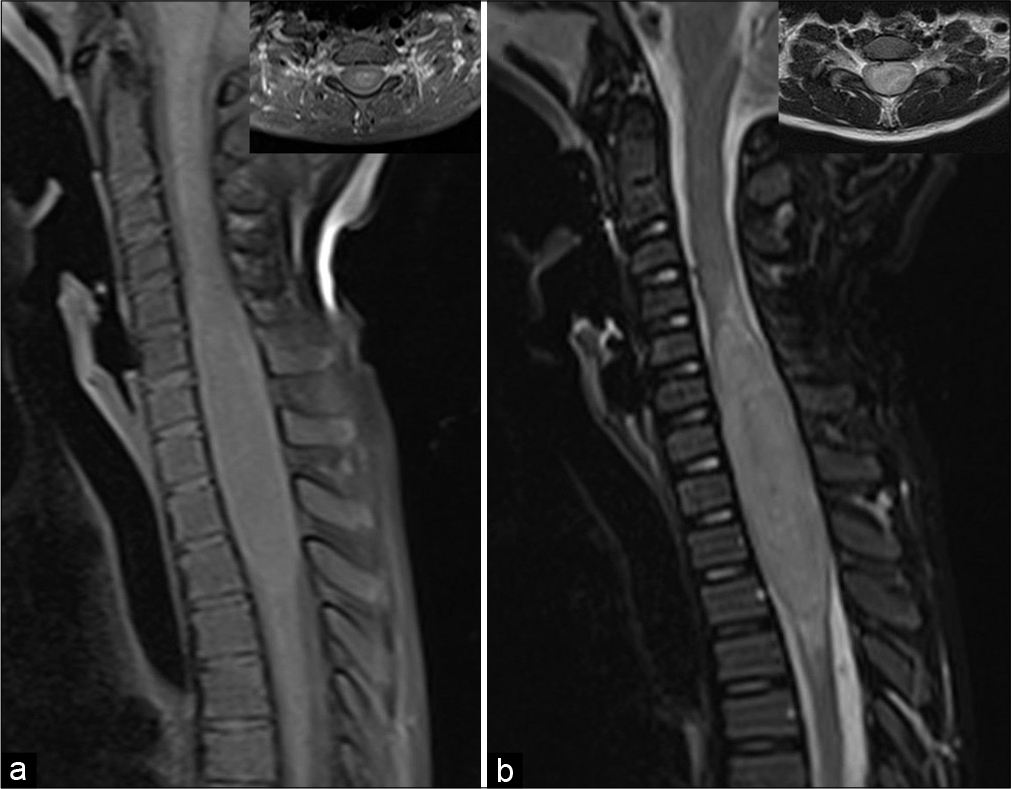
Figure 1:
Cervicothoracic spinal magnetic resonance imaging. (a) Axial and sagittal views of contrast-enhanced T1-weighted with fat suppression demonstrated an intramedullary lesion from C4 to T4. Note the peripheral isointense signal with an irregular central hyperintense component. (b) The lesion was hyperintense on short-time inversion recovery (sagittal slice) and T2-weighted (axial slice) sequences.
After initial evaluation, it was decided to perform a cervicothoracic tumor resection. The procedure was performed under intravenous general anesthesia. After anesthetic induction, the patient was placed in the prone position with a 3-pin head holder. Antisepsis and asepsis techniques were performed with subsequent placement of sterile drapes. A posterior longitudinal skin incision was made. After identification, through palpation and guided radioscopy of the cervical and thoracic spinous processes, a subperiosteal dissection was performed. The patient underwent a C3-T5 laminotomy. After bone removal, the dura mater was opened and attached with 4-0 polypropylene suture. During inspection, the tumor was intramedullary and situated laterally (left side) in the cervicothoracic spinal cord. The lesion consistency was apparently firm, with a good cleavage plane in the superior aspect of the spinal cord [
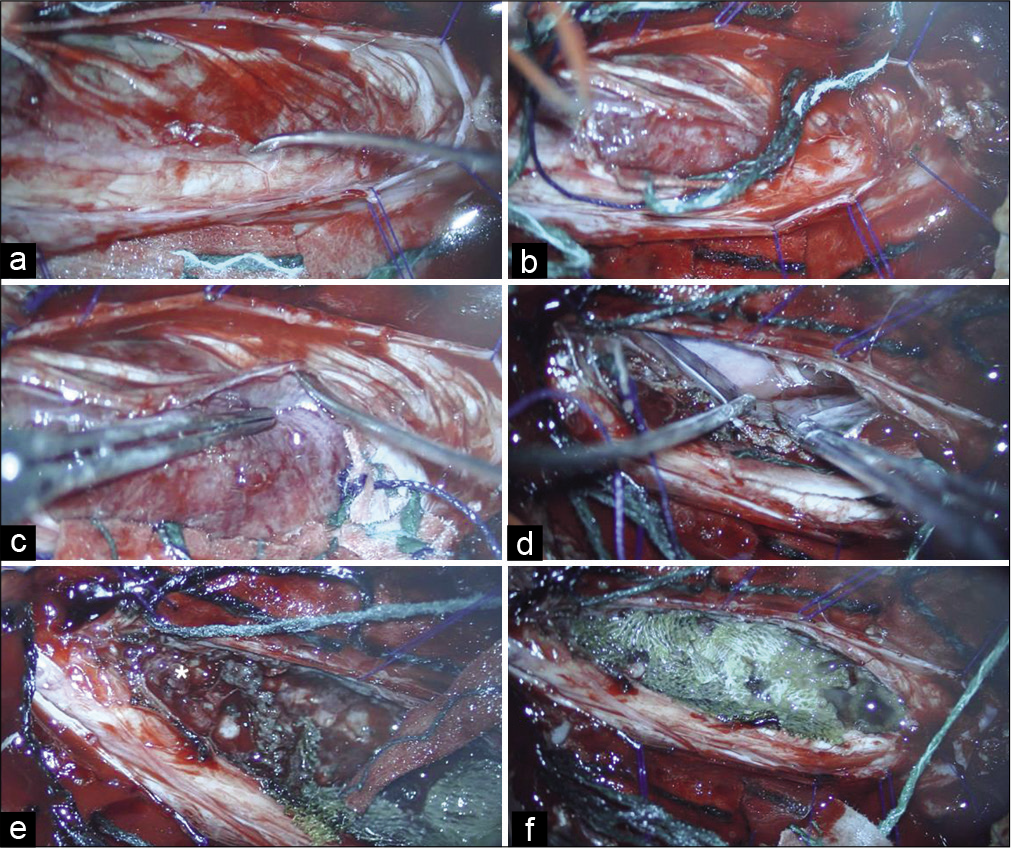
Figure 2:
Microsurgery resection of the cervicothoracic tumor. (a) The lesion was located laterally (left side) pushing the dorsal cervical nerve roots (arrow head). (b) Some dorsal nerve roots were removed to allow lesion removal. (c) Tumor dissection included the most lateral aspect. (d) Subarachnoid plane after lateral dissection. (e) The most inferior aspect of the tumor was very attached. Traction and dissection of this part of the tumor cause a significant drop of the intraoperative somatosensory and motor evoked potentials. It was decided to leave some tumor (asterisk). (f) Final aspect of the cavity. The tumor bed was filled with absorbent cellulose hemostatics.
After the initial dissection of the upper pole of the tumor, partial resection of the lesion was performed. In the most caudal portion, the tumor was infiltrating the adjacent neural tissue without a clear cleavage plane. On the basis of a drop of the intraoperative somatosensory and motor evoked potentials (MEPs) during dissection, we choose to leave some tumor. Therefore, a partial resection was achieved [
During the procedure, the somatosensory evoked potentials recorded cortical waves P37 and N20, with stable latencies and amplitudes. There was a MEP amplitude decreasing (90%) of the triceps brachii bilaterally and left abductor pollicis brevis after complete removal of the superior tumor pole [
Figure 3:
Intraoperative neurophysiological monitoring – right side. (a) Demonstration of motor evoked potential (MEP) with stable amplitude and latency at the beginning of the procedure (gray circle). The following muscles were analyzed: deltoid, triceps brachii, abductor pollicis brevis, tibialis anterior, and abductor hallucis. (b) After superior tumor pole removal, there was a MEP drop in the right triceps brachii muscle (green circle). There were no changes in the other muscles MEPs during inferior pole dissection. (c) At the end of the procedure, there was a MEP normalization on the right side (blue circle).
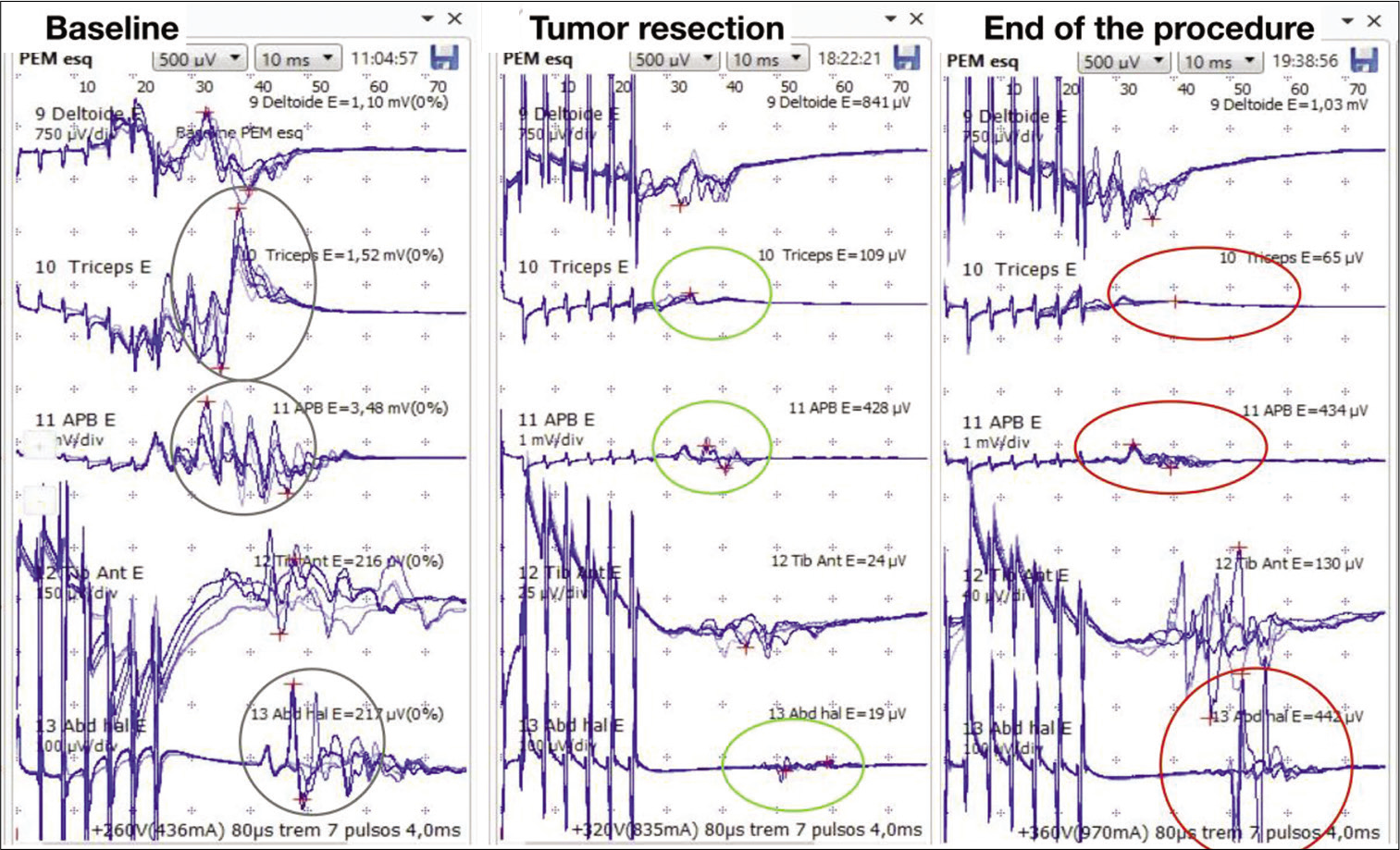
Figure 4:
Intraoperative neurophysiological monitoring – left side. (a) Demonstration of motor evoked potential (MEP) with stable amplitude and latency at the beginning of the procedure (gray circle). The following muscles were analyzed: deltoid, triceps brachii, abductor pollicis brevis, tibialis anterior, and abductor hallucis. (b) After superior tumor pole removal, there was 90% MEP drop in the left triceps brachii and abductor pollicis brevis (green circle). During inferior resection, there was 89% MEP drop in the left abductor hallucis and the resection was interrupted (green circle). (c) At the end of the procedure, the MEP signal drop in the left triceps brachii and abductor pollicis brevis was maintained (red circle). The left abductor hallucis MEP returned to the baseline (red circle).
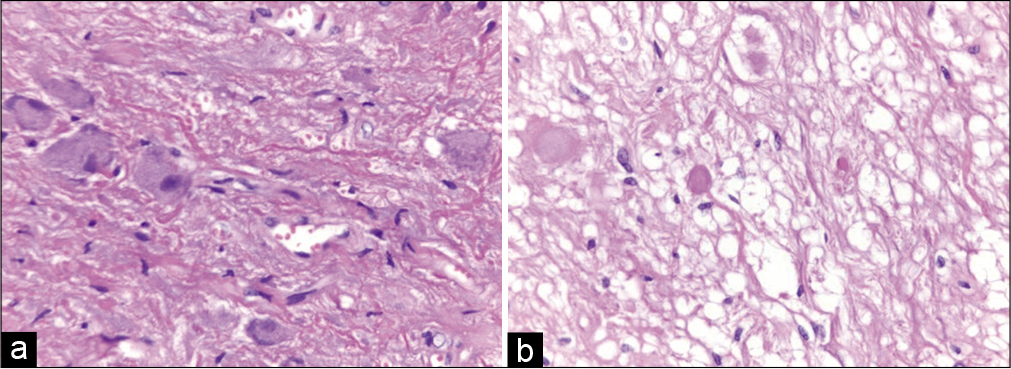
Histopathological examination revealed a relative well-circumscribed neoplasm composed of atypical neurons distributed irregularly. The neurons were mixed with glial proliferation, mild atypia, and a dense fibrillary background with focal desmoplasia. There were eosinophilic granular bodies and rare microcalcification besides vessels with hyalinized wall. These findings supported the diagnosis of ganglioglioma – World Health Organization Grade I [
In the immediate postoperative period, the patient presented kinesiophobia, refusing to get out of bed in the 1st days. Worsening of paraparesis was noted (Grade 3 MRC bilaterally). The patient did not use steroids in the pre- and post-operative period. The residual lesion size measured by the immediate postoperative spine MRI was 1.6 × 1.5 × 1.3 cm (length; depth; and width) representing 80% of tumor resection. The patient was discharged and returned 3 weeks after the procedure. He presented a symmetrical Grade 4 MRC paraparesis with spasticity, hyperreflexia, and bilateral Babinski signs. He improved his preoperative functional neurological state exhibiting a spastic gait without fatigue overtime. He is being followed up for 7 months with serial clinical and radiological examinations. In the last appointment, there was a significant improvement in gait (Grade 5 MRC scale on the right leg and Grade 4+ on the left leg). Postoperative MRI, performed 5 months after surgery, showed no growth of residual lesion [
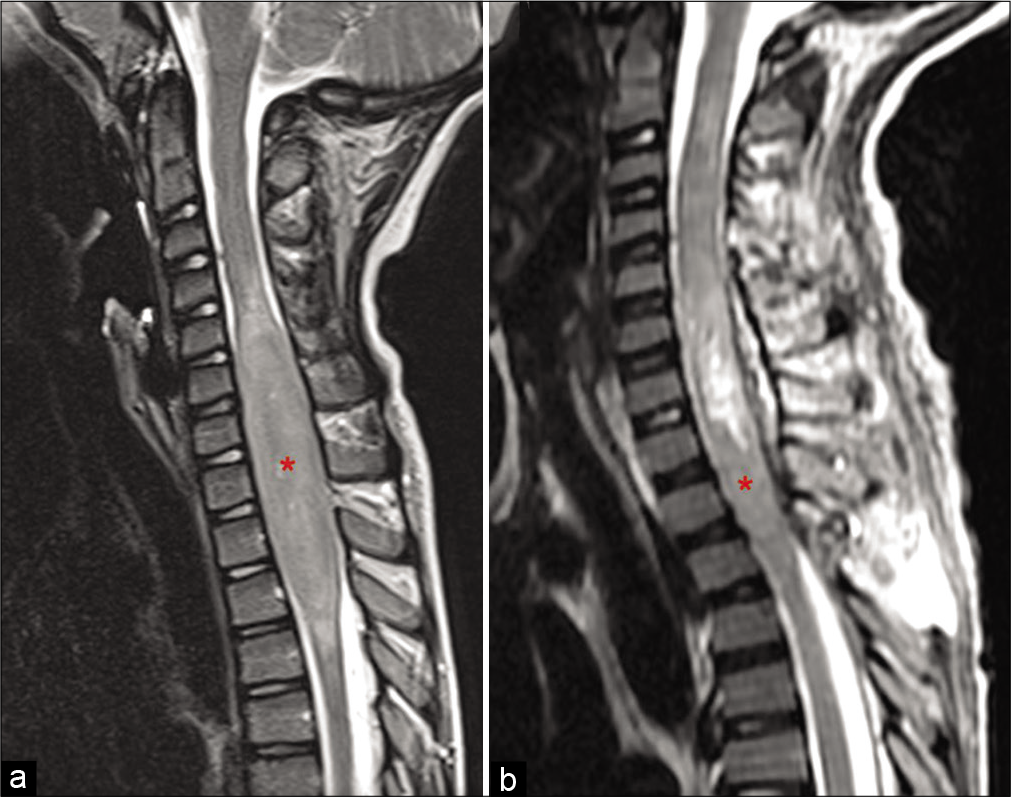
Figure 6:
Comparison between pre- and post-operative cervicothoracic spine magnetic resonance imaging. (a) Preoperative sagittal view of T2-weighted demonstrating an intramedullary lesion from C4 to T4 (red asterisk). (b) Postoperative sagittal view of T2-weighted demonstrating the hyperintense residual cavity and caudal residual tumor (red asterisk).
DISCUSSION
Spinal cord gangliogliomas are very rare tumors. A study involving 348 children, demonstrated a preference for males and an average of 10 years old.[
Malignant progression of gangliogliomas is a rare event, reported in the literature in 10% of cases. It is considered that the proliferation of the astrocytic component is responsible for this malignant transformation, while the neuronal component does not have a neoplastic nature.[
About 40% of patients with NF1 have spinal cord lesions on MRI. They lead to clinical manifestations in approximately 2% of patients.[
Due to the rarity of the association of ganglioglioma and NF1, little is known about the incidence, age of presentation, sex preference, most common location in the spinal cord, best treatment, and prognosis. Despite the total resection of spinal gangliogliomas being the standard treatment, in this case, we were not able to perform a complete resection because the MEP dropped during caudal dissection. The tumor was infiltrating the spinal cord. To preserve neurofunctional status, it was chosen to leave some tumor and follow-up the residual lesion. Its indolent tumor nature, allows an expectant approach. It is important to distinguish gangliogliomas from the other common NF1-associated tumors, especially diffuse astrocytoma, pilocytic astrocytoma, and focal areas of signal intensity.
CONCLUSION
This report adds to the literature another case of a spinal ganglioglioma in a patient with NF1. The tumor must be included in the differential diagnosis of patients with NF1 and spinal lesions. Despite complete microsurgery resection being the standard treatment of gangliogliomas, in this case, it was decided to perform a partial resection and follow-up due to the high risk of spinal cord injury. The prognosis and treatment of this condition associated with NF1 remains to be determined.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Center of High Complexity Neurosurgery Intern Patients (NIPNAC).
Conflicts of interest
There are no conflicts of interest.
References
1. Araújo JF, Souza MR, Sperlescu A, Balbo RJ. Malignant course of a ganglioglioma: Case report. Arq Neuropsiquiatr. 1998. 56: 486-90
2. Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A. Recurrent mutations in the NF1 gene are common among neurofibromatosis Type 1 patients. J Med Genet. 2003. 40: e82
3. Cawthon RM, O’Connell P, Buchberg AM, Viskochil D, Weiss RB, Culver M. Identification and characterization of transcripts from the neurofibromatosis 1 region: The sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990. 7: 555-65
4. Dudley RW, Torok MR, Gallegos DR, Mulcahy-Levy JM, Hoffman LM, Liu AK. Pediatric low-grade ganglioglioma: Epidemiology, treatments, and outcome analysis on 348 children from the surveillance, epidemiology, and end results database. Neurosurgery. 2015. 76: 313-9
5. Fedi M, Mitchell LA, Kalnins RM, Gutmann DH, Perry A, Newton M. Glioneuronal tumours in neurofibromatosis Type 1: MRI-pathological study. J Clin Neurosci. 2004. 11: 745-7
6. Giussani C, Isimbaldi G, Massimino M, Trezza A, Cianci P, Conocico F. Ganglioglioma of the spinal cord in neurofibromatosis Type 1. Pediatr Neurosurg. 2013. 49: 50-4
7. Gorlin RJ, Cohen MM, Levin LS.editors. Hamartoneoplastic syndromes. Syndromes of the Head and Neck. Oxford: Oxford University Press; 1990. p. 353-416
8. Gupta K, Orisme W, Harreld JH, Qaddoumi I, Dalton JD, Punchihewa C. Posterior fossa and spinal gangliogliomas form two distinct clinicopathologic and molecular subgroups. Acta Neuropathol Commun. 2014. 2: 18
9. Hayashi Y, Nakada M, Mohri M, Murakami H, Kawahara N, Hamada J. Ganglioglioma of the thoracolumbar spinal cord in a patient with neurofibromatosis Type 1: A case report and literature review. Pediatr Neurosurg. 2011. 47: 210-3
10. Jallo G, Freed D, Epstein F. Spinal cord gangliogliomas: A review of 56 patients. J Neurooncol. 2004. 68: 71-7
11. Junior L, Filho A, Silva D, Geller M. Neurofibromatosis type 1 in childhood: Review of clinical aspects. Rev Paul Pediatr. 2008. 26: 176-82
12. Lang FF, Epstein FJ, Ransohoff J, Allen JC, Wisoff J, Abbott IR. Central nervous system gangliogliomas Part.2 Clinical outcome. J Neurosurg. 1993. 79: 867-73
13. Lee M, Rezai AR, Freed D, Epstein FJ. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996. 38: 32-7
14. Miller DC. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000. 47: 189-94
15. Patel U, Pinto RS, Miller DC, Handler MS, Rorke LB, Epstein FJ. MR of spinal cord ganglioglioma. AJNR Am J Neuroradiol. 1998. 19: 879-87
16. Patibandla MR, Ridder T, Dorris K, Torok MR, Liu AK, Handler MH. Atypical pediatric ganglioglioma is common and associated with a less favorable clinical course. J Neurosurg Pediatr. 2016. 17: 41-8
17. Rodriguez FJ, Perry A, Gutmann DH, O’Neill BP, Leonard J, Bryant S. Gliomas in neurofibromatosis Type 1: A clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008. 67: 240-9
18. Silver JM, Rawlings CE, Rossich E, Zeidman SM, Friedman AH. Ganglioglioma: A clinical study with long-term follow-up. Surg Neurol. 1991. 35: 261-6
19. Sundar IV, Jaiswal M, Purohit D, Mittal RS. Ganglioglioma of conus medullaris in a patient of neurofibromatosis type 1: A novel association?. Asian J Neurosurg. 2016. 11: 455
20. Tian R, Liu B, Wang G, Hao S. Natural history of spinal ganglioglioma in neurofibromatosis Type 1: A 10-year follow-up. Int J Clin Exp Med. 2016. 9: 4879-83
21. Truite LV, Hanel RA, Grande CV, Torres LF, Aráujo JC. Spinal cord ganglioglioma: Case report. Arq Neuropsiquiatr. 2001. 59: 431-4
22. Zhang M, Iyer RR, Azad TD, Wang Q, Garzon-Muvdi T, Wang J. Genomic landscape of intramedullary spinal cord gliomas. Sci Rep. 2019. 9: 18722

