

- Department of Neurosurgery, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, New York, United States.
- Department of Oral and Maxillofacial Surgery, University at Buffalo School of Dental Medicine, Buffalo, New York, United States.
- Department of Pediatrics, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, New York, United States.
Correspondence Address:
Matthew J. Recker, Department of Neurosurgery, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, New York, United States.
DOI:10.25259/SNI_329_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Matthew J. Recker1, Nathaniel Kronenwetter2, Renée M. Reynolds1, Laurie S. Sadler3, Michael R. Markiewicz2. Impaired wound healing following cranial vault reconstruction in a patient with an atypical phenotype of Marfan syndrome: A case report. 29-Jul-2022;13:328
How to cite this URL: Matthew J. Recker1, Nathaniel Kronenwetter2, Renée M. Reynolds1, Laurie S. Sadler3, Michael R. Markiewicz2. Impaired wound healing following cranial vault reconstruction in a patient with an atypical phenotype of Marfan syndrome: A case report. 29-Jul-2022;13:328. Available from: https://surgicalneurologyint.com/surgicalint-articles/11750/
Date of Submission
08-Apr-2022
Date of Acceptance
12-Jul-2022
Date of Web Publication
29-Jul-2022
Abstract
Background: Marfan syndrome (MFS) is an autosomal dominant disorder of the connective tissues caused by mutations in the FBN1 gene which can result in widespread systemic involvement. Loeys-Dietz syndrome (LDS) is a related autosomal dominant disorder of connective tissue with widespread systemic involvement which has phenotypic overlap with MFS. LDS is caused by heterozygous pathogenic variants in six different genes, the most common of which involve transforming growth factor beta-receptor 1 or 2. While LDS is commonly associated with craniofacial manifestations, MFS is not typically characterized by craniosynostosis.
Case Description: We present a 7-month-old female patient with MFS and metopic craniosynostosis with an unusual clinical presentation who underwent cranial vault reconstruction with fronto-orbital advancement and anterior cranial vault remodeling. Her course was complicated by impaired wound healing after surgery, requiring return to the operating room.
Conclusion: Phenotypic overlap between genetic disorders can confound clinical diagnosis as illustrated in this case. Genetic testing can be highly valuable in the diagnosis of clinically variable disorders. Patients with MFS who undergo cranial surgery may be at increased risk for wound healing complications.
Keywords: Craniofacial abnormalities, Craniofacial dysostosis, Craniosynostosis, Marfan syndrome, Pediatrics
INTRODUCTION
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder that is characterized by varying combinations of skeletal, cardiovascular, pulmonary, dermatologic, and ocular malformations.[
LDS is a related autosomal dominant disorder of connective tissue with widespread systemic involvement. It is caused by heterozygous pathogenic variants in six different genes, the most common of which are transforming growth factor beta-receptor 1 or 2 (TGFBR1 or TGFBR2).[
CASE DESCRIPTION
A 7-month-old female presented to the Craniofacial Center of Western New York (John R. Oishei Children’s Hospital, Buffalo, NY) following a genetics consultation that raised concern for LDS with possible craniosynostosis. The parent’s chief complaint was the appearance of the patient’s head shape and orbits, which they felt had been abnormal since birth. The parent’s denied any constitutional symptoms. They denied any apparent visual disturbances, abnormal gaze, signs of headache, head banging, unexplained vomiting, or other symptoms classically associated with increased intracranial pressure. The patient was noted to be tracking above average on her growth curve and was noted to be meeting all developmental milestones. Physical exam revealed a well-appearing, tall infant. She had a pectus excavatum deformity, arachnodactyly, and long narrow feet with long toes. Her craniofacial examination was remarkable for ridging along the metopic and left coronal sutures with bitemporal narrowing and mild trigonocephaly, a flat anterior fontanel, and prominent supraorbital ridges [
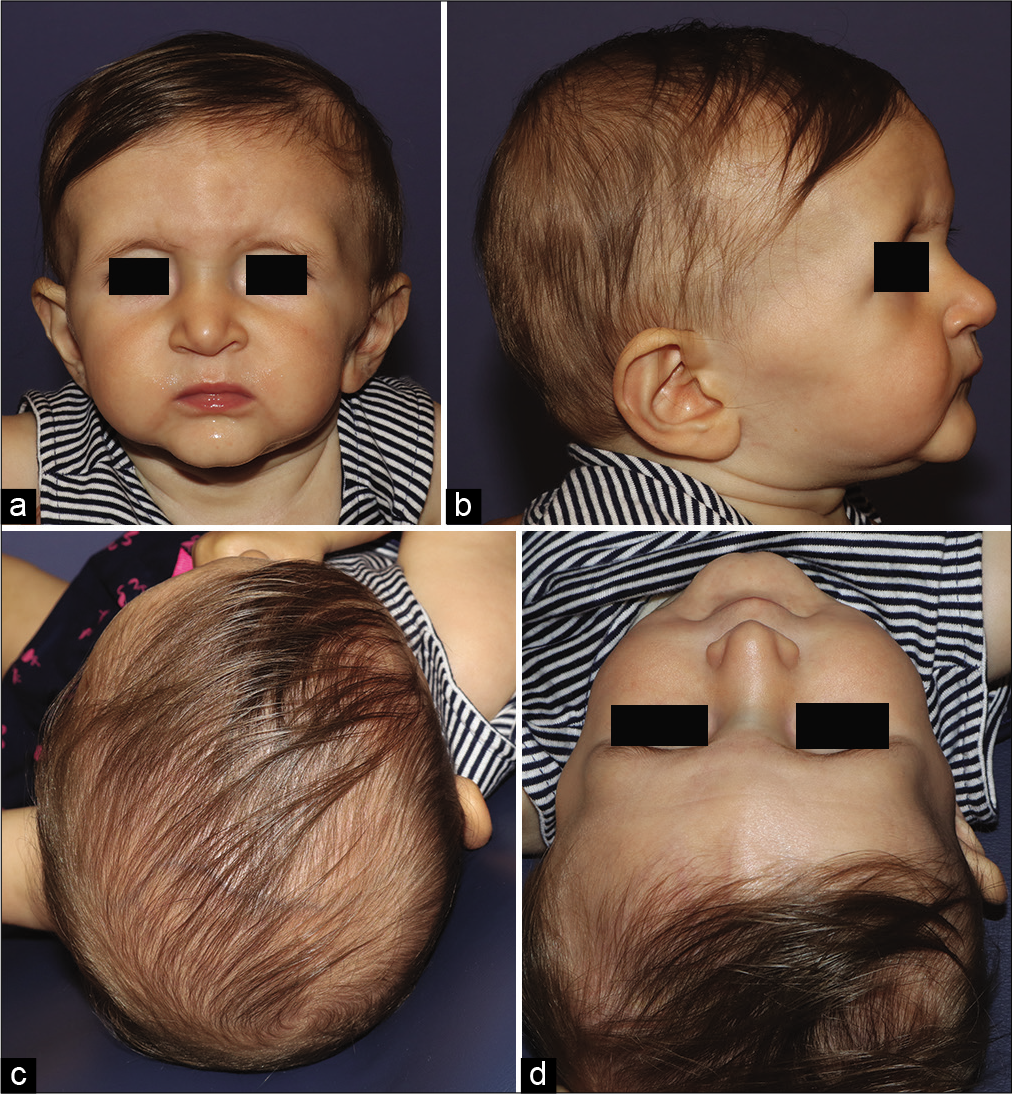
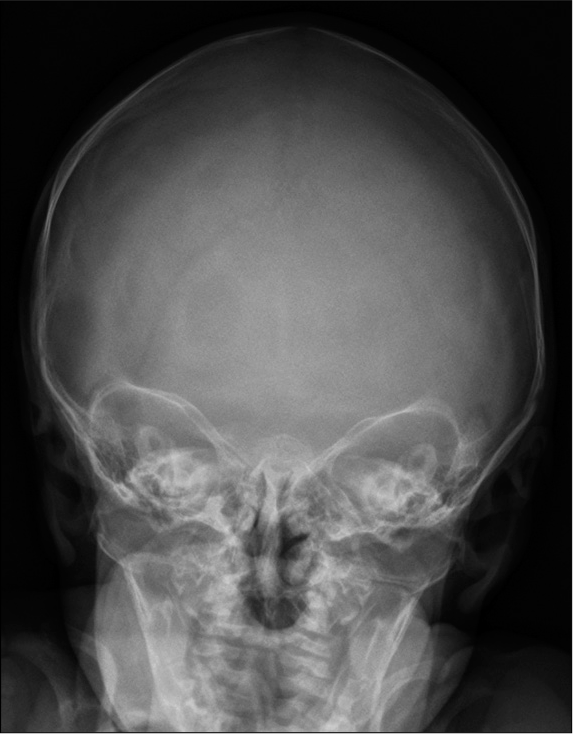
Although the patient had presented to the Craniofacial Clinic at 7 months of age, there was concern for the child’s abnormal head shape early in life and on presenting to the child’s physician, a posterior-anterior skull radiograph was taken at 7 weeks of age. The skull radiograph skull radiograph identified metopic suture craniosynostosis [
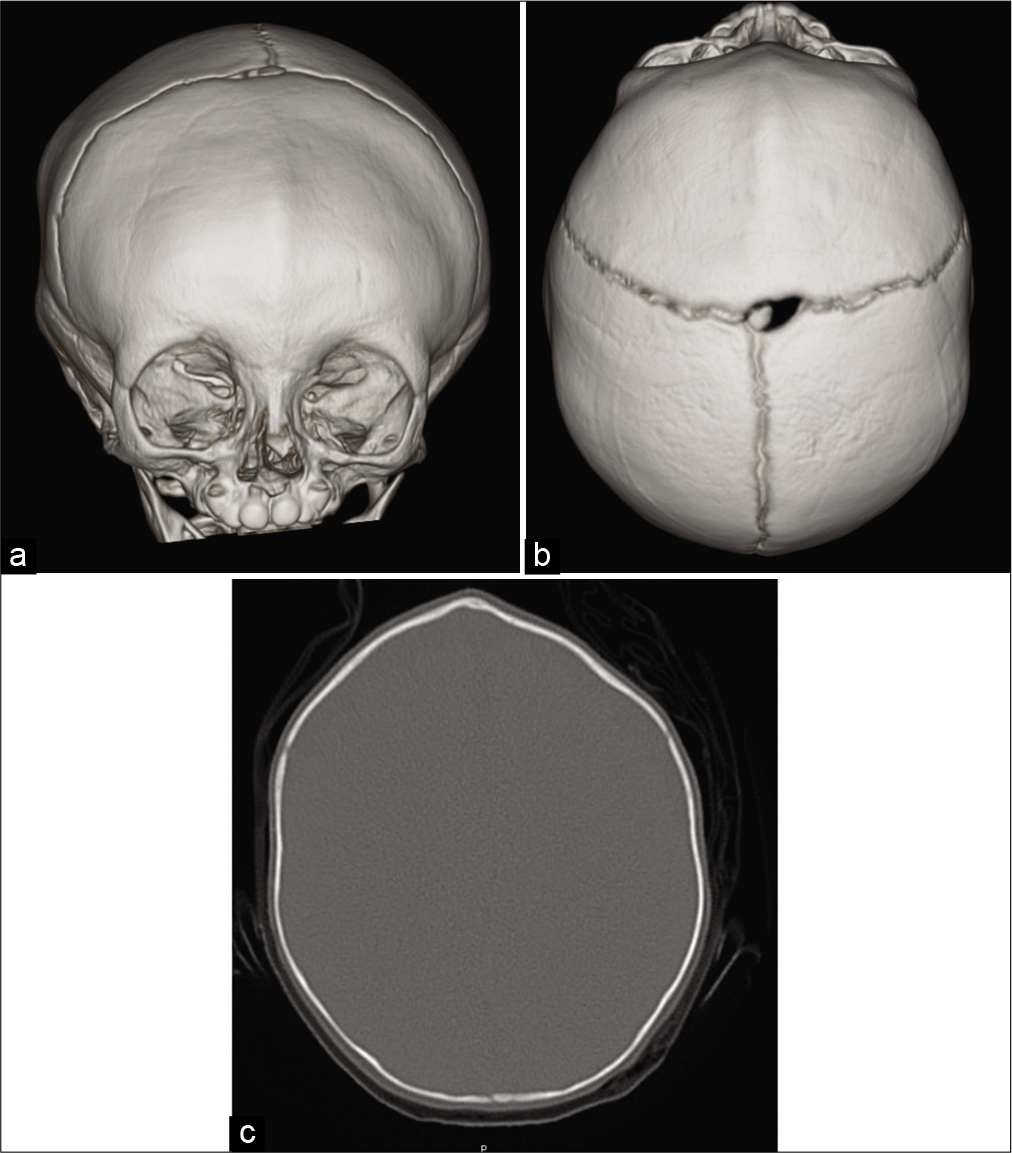
The patient was taken to the operating room at age 9 months for fronto-orbital advancement and anterior cranial vault expansion and remodeling. A standard curved coronal incision extending bilaterally to 2 cm superior of the postauricular area was made. An avascular subgaleal was then dissected up to 1.5 cm superior to the supraorbital rims. Next, an anteriorly based pericranial flap exposing the frontonasal suture and frontozygomatic sutures was dissected in the subperiosteal plane [
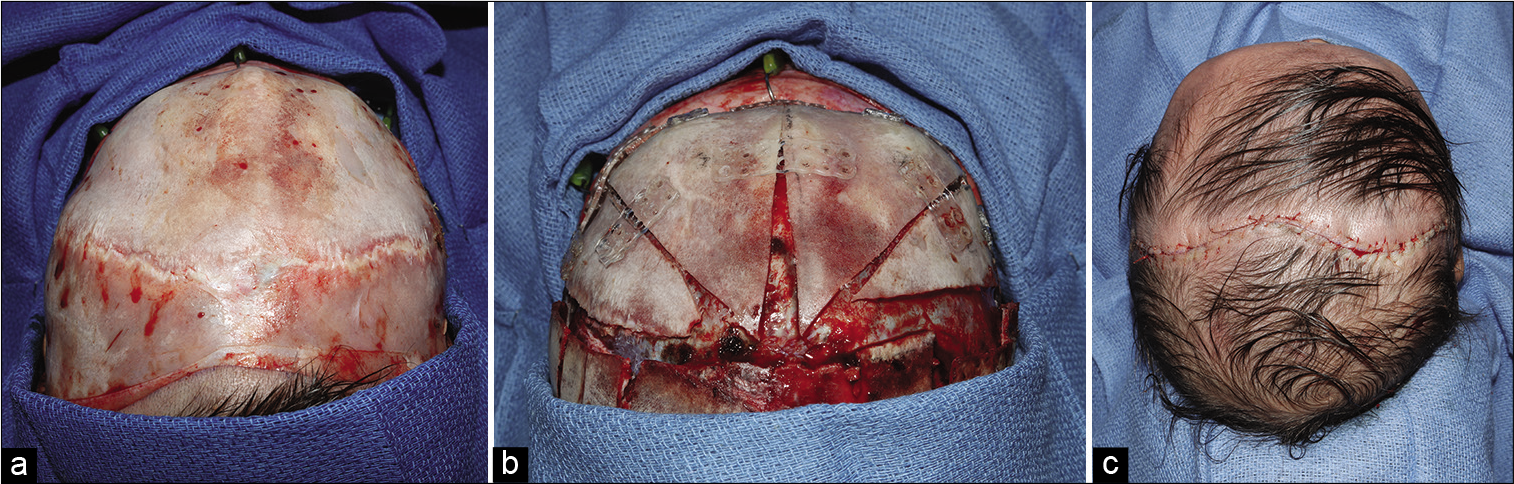
The patient followed up 3 days later (postoperative day 7) displaying adequate head shape and good wound healing. Her scalp incision appeared well approximated with no signs of infection or dehiscence [
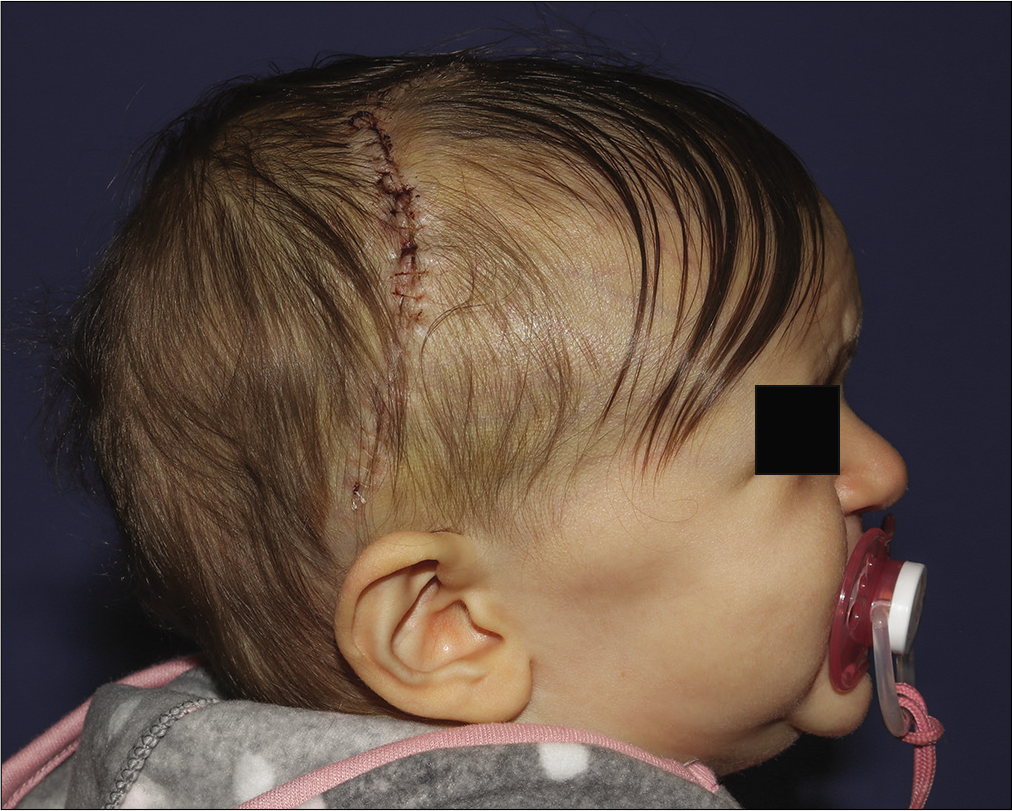
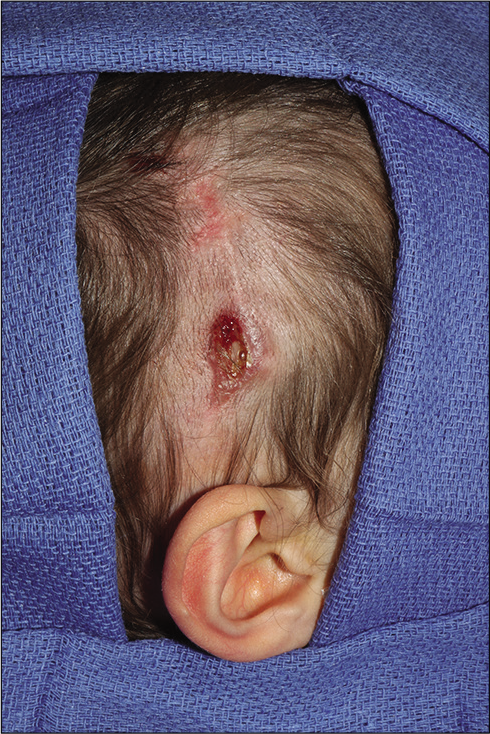
The patient underwent genetic testing which disclosed a variant in a highly conserved region of the FBN1 gene denoted as c.3152T>C(p.phe1051ser). This variant is classified as a variant of unknown significance, but this mutation in combination with the patient’s phenotype was considered to be pathogenic and diagnostic of MFS. There were no variants detected in the other six genes analyzed. Results of follow-up parental studies were negative for the FBN1 variant detected in our patient, confirming that the variant was de novo.
DISCUSSION
We present a case of MFS and metopic craniosynostosis with notable atypical findings. First, our patient’s clinical phenotype suggested a diagnosis of LDS. Her genetic test results, however, were compatible with a diagnosis of MFS. Our patient’s craniofacial findings, particularly her craniosynostosis, confounded our initial diagnostic impression. Although there is currently no standard or widely accepted craniometric measurement for the diagnosis of metopic craniosynostoses, several have been proposed in the contemporary literature.[
Ozyurt et al. reported two cases of early onset MFS.[
These cases, in conjunction with the case presented herein, further demonstrate the wide clinical spectrum of MFS. There appears to be a phenotypic continuum among patients clinically diagnosed with MFS, marfanoid features-craniosynostosis, or marfanoid features-mental retardation spectrum. The underlying genetic variants in patients with MFS who have craniofacial abnormalities are likely related to abnormal TGF-b signaling, affecting craniofacial morphogenesis. Our findings indicate that craniosynostosis does not exclude a diagnosis of MFS, although it is not a typical finding.
Another interesting aspect of the patient in this report is her poor postoperative healing. It is reported in the general surgery literature that patients with MFS tend to develop hernias, which can be difficult to repair, because the soft tissue can fail to retain sutures.[
Classical Ehlers–Danlos syndrome (EDS), a disorder affecting collagen structure and processing, is characterized by abnormal wound healing. Affected patients demonstrate skin fragility and often experience delayed wound healing. Surgical recommendations include that dermal wounds be closed in at least two layers, and sutures should be left in place twice as long as usual. In addition, some recommend reinforcement of the wound with adhesive tape.[
Van Camp et al. performed a large retrospective study of patients with classical EDS and MFS who presented to the Oral and Maxillofacial surgery department at their institution.[
CONCLUSION
Phenotypic overlap between genetic disorders can confound clinical diagnosis, as illustrated in this case. Genetic testing can be highly valuable in the diagnosis of clinically variable disorders. Patients with MFS who undergo cranial surgery may be at increased risk for wound healing complications.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ades LC, Sullivan K, Biggin A, Haan EA, Brett M, Holman KJ. FBN1, TGFBR1, and the Marfan-craniosynostosis mental retardation disorders revisited. Am J Med Genet A. 2006. 140: 1047-58
2. Beyene RT, Derryberry SL, Barbul A. The effect of comorbidities on wound healing. Surg Clin North Am. 2020. 100: 695-705
3. Chandler L, Park KE, Allam O, Mozaffari MA, Khetpal S, Smetona J. Distinguishing craniomorphometric characteristics and severity in metopic synostosis patients. Int J Oral Maxillofac Surg. 2021. 50: 1040-6
4. Gray JR, Davies SJ. Marfan syndrome. J Med Genet. 1996. 33: 403-8
5. McEwan TW, Martin AL, Tanaka T, Aldridge K, Muzaffar AR. Evaluating children with metopic craniosynostosis: The cephalic width-intercoronal distance ratio. Cleft Palate Craniofac J. 2016. 53: e95-100
6. Naran S, Mazzaferro D, Wes A, Vossough A, Bartlett SP, Taylor JA. A craniometric analysis of cranial base and cranial vault differences in patients with metopic craniosynostosis. J Craniofac Surg. 2017. 28: 2030-5
7. Ozyurt A, Baykan A, Argun M, Pamukcu O, Halis H, Korkut S. Early onset marfan syndrome: A typical clinical presentation of two cases. Balkan J Med Genet. 2015. 18: 71-6
8. Sood S, Eldadah ZA, Krause WL, McIntosh I, Dietz HC. Mutation in fibrillin-1 and the marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat Genet. 1996. 12: 209-11
9. Teager SJ, Constantine S, Lottering N, Anderson PJ. Physiologic closure time of the metopic suture in South Australian infants from 3D CT scans. Childs Nerv Syst. 2019. 35: 329-35
10. Tsang AK, Taverne A, Holcombe T. Marfan syndrome: A review of the literature and case report. Spec Care Dentist. 2013. 33: 248-54
11. Van Camp N, Aerden T, Politis C. Problems in the orofacial region associated with Ehlers-Danlos and marfan syndromes: A case series. Br J Oral Maxillofac Surg. 2020. 58: 208-13
12. Van Hemelrijk C, Renard M, Loeys B. The loeys-dietz syndrome: An update for the clinician. Curr Opin Cardiol. 2010. 25: 546-51
13. Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan syndrome and related disorders: 25 Years of gene discovery. Hum Mutat. 2016. 37: 524-31
14. Vu HL, Panchal J, Parker EE, Levine NS, Francel P. The timing of physiologic closure of the metopic suture: A review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J Craniofac Surg. 2001. 12: 527-32
15. Wagner AH, Zaradzki M, Arif R, Remes A, Muller OJ, Kallenbach K. Marfan syndrome: A therapeutic challenge for long-term care. Biochem Pharmacol. 2019. 164: 53-63
16. Wang JY, Dorafshar AH, Liu A, Groves ML, Ahn ES. The metopic index: An anthropometric index for the quantitative assessment of trigonocephaly from metopic synostosis. J Neurosurg Pediatr. 2016. 18: 275-80
17. Weinzweig J, Kirschner RE, Farley A, Reiss P, Hunter J, Whitaker LA. Metopic synostosis: Defining the temporal sequence of normal suture fusion and differentiating it from synostosis on the basis of computed tomography images. Plast Reconstr Surg. 2003. 112: 1211-8

