

- Departments of Neurosurgery, St George Hospital, Kogarah, Australia,
- Anatomical Pathology, St George Hospital, Kogarah, Australia.
Correspondence Address:
Cher Shui, Department of Neurosurgery, St George Hospital, Kogarah, Australia.
DOI:10.25259/SNI_31_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Cher Shui1, Louella Davey2, Martin Scholsem1. Leptomeningeal dissemination of a malignant melanotic nerve sheath tumor: A case report and review of the literature. 18-Feb-2022;13:59
How to cite this URL: Cher Shui1, Louella Davey2, Martin Scholsem1. Leptomeningeal dissemination of a malignant melanotic nerve sheath tumor: A case report and review of the literature. 18-Feb-2022;13:59. Available from: https://surgicalneurologyint.com/surgicalint-articles/11400/
Date of Submission
08-Jan-2022
Date of Acceptance
24-Jan-2022
Date of Web Publication
18-Feb-2022
Abstract
Background: Malignant melanotic nerve sheath tumors (MMNSTs) are rare tumors of presumed neural crest origin. Here, we present a 21-year-old female with a left L5/S1 MMNST along with a review of approximately 70 spinal cases reported in the literature, the majority of which were either local recurrences or metastases.
Case Description: A 21-year-old female presented with 3 months of severe left L5 distribution radicular leg pain and sensory loss. The MR revealed a dumbbell-shaped, heterogenously enhancing lesion centered on the left L5/S1 foramen; the intracanalicular component displaced the thecal sac to the right, while the extraforaminal portion of tumor extended anteriorly into the retroperitoneal space. Gross total resection was performed after a L5/S1 facetectomy. In the immediate postoperative period there were no complications, and the patient had full lower limb power. Four months later, the patient experienced generalized seizures, headache, and multiple cranial nerve palsies due to local and diffuse CNS dissemination. The MRI of the brain and whole spine revealed diffuse leptomeningeal enhancement along the full length of the spinal cord into the brainstem and cerebrum along with a focally recurrent epidural soft-tissue lesion located posterolaterally on the left at the L4/5 level (i.e., measuring 12 mm × 10 mm). An external ventricular drain and subsequent ventriculoperitoneal shunt were inserted, followed by craniospinal irradiation. She was discharged 3 months later with residual distal lower limb weakness.
Conclusion: This case illustrates the rapid disease progression of MMNST despite gross total resection. Further such lesions should be aggressively treated locally, and followed by adjuvant radiotherapy and systemic chemotherapy/immunotherapy.
Keywords: Adjuvant radiotherapy, Leptomeningeal dissemination, Malignant melanotic nerve sheath tumor, Melanotic schwannoma, Recurrence
INTRODUCTION
Malignant melanotic nerve sheath tumors (MMNSTs) of the spine are rare, and fewer than 70 cases are described in the literature.[
CASE REPORT
Original presentation and surgery
A 21-year-old female presented with 3 months of severe, left L5 radicular pain accompanied by L5 hypoesthesia to light touch and decreased pin-perception, without weakness. MR showed a dumbbell-shaped, heterogenously enhancing lesion centered in the left L5/S1 foramen, measuring 4.5 cm × 4 cm [
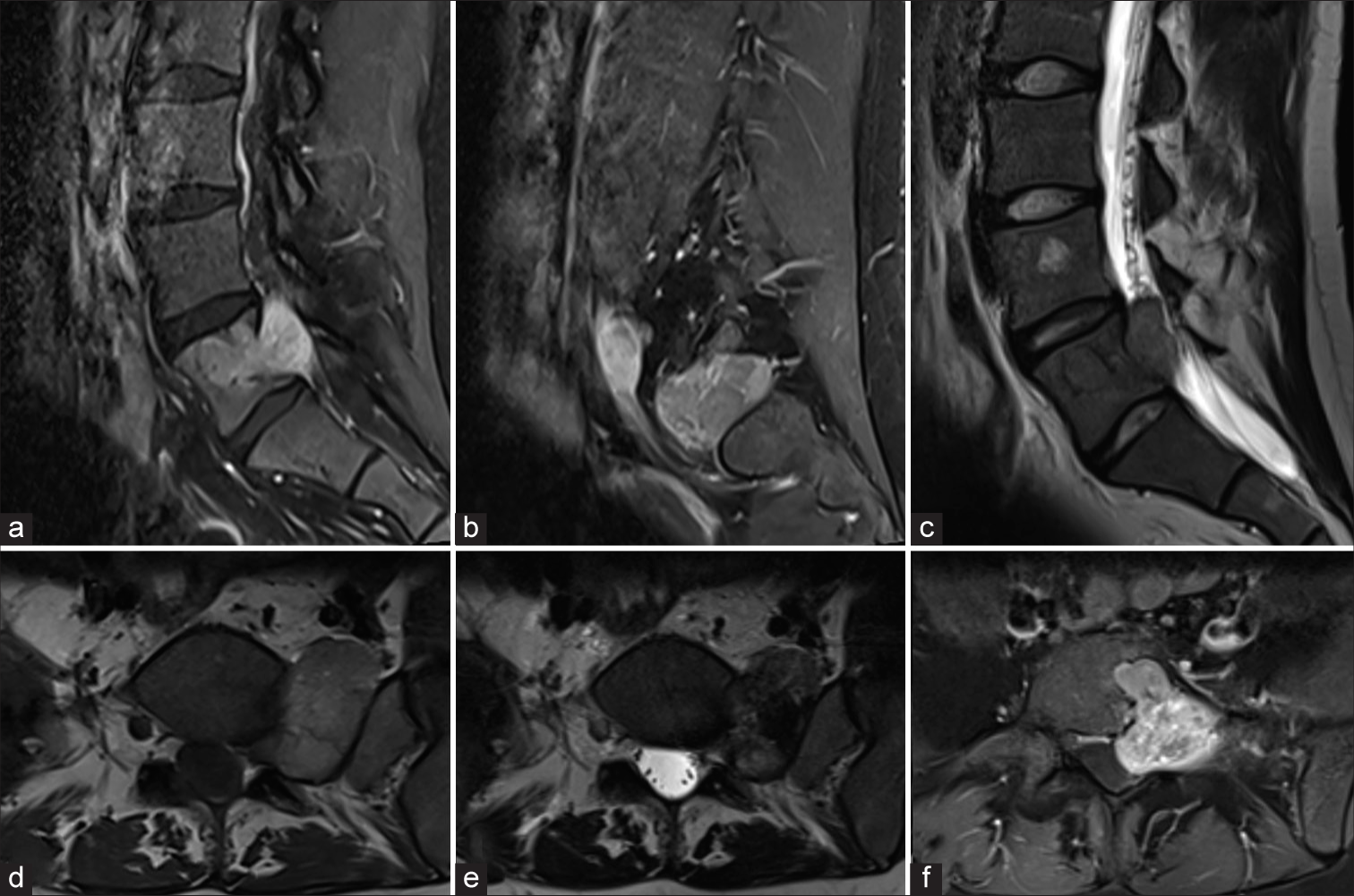
Figure 1:
Preoperative MR images of the lesion. (a and b) Sagittal T1-weighted images with gadolinium enhancement of the lumbosacral spine demonstrating diffuse, heterogenous enhancement of the lesion with involvement the posterior aspect of the L5 vertebrae extending laterally to the retroperitoneal space. (c) Sagittal T2-weighted MR image showing extension of the lesion into the spinal canal and incidental hemangioma in the L4 vertebral body. (d) Axial T1-weighted MR image with lesion exiting the left L5/S1 foramen. (e) Axial T2-weighted MR image with lesion demonstrating low signal. (f) Axial T1-weighted image of the lesion with gadolinium enhancement showing heterogenous enhancement and rightward displacement of the thecal sac.
Operation
Through a L4 and L5 hemilaminectomy with a left L5/S1 facetectomy, a gross total excision was accomplished. On opening of the dura, a black, hemorrhagic tumor was visualized. Postoperatively, the patient’s radicular pain resolved, she retained full motor and regained normal sensory function in the left L5 distribution.
Local recurrence and leptomeningeal spread of tumor
Within 4 postoperative months and despite a negative PETCT obtained 3 months after surgery, the lesion recurred. The patient presented with sudden onset lower back pain with recurrent radiculopathy. Within days, she also developed a severe bifrontal headache, vomiting, photophobia, visual loss (i.e., only able to differentiate between light and dark), and a generalized seizure. The patient also developed distal lower limb weakness, bilateral abducens nerve palsies, facial diplegia, bilateral trigeminal nerve palsies, and dysarthria. The fundus exam showed florid papilledema. The emergent holo-spinal and brain MRI showed diffuse leptomeningeal enhancement along the full length of the spinal cord extending to the brainstem and cerebrum. There was also a focal recurrent epidural soft-tissue lesion within the left posterolateral aspect at L4/5 measuring 12 mm × 10 mm [
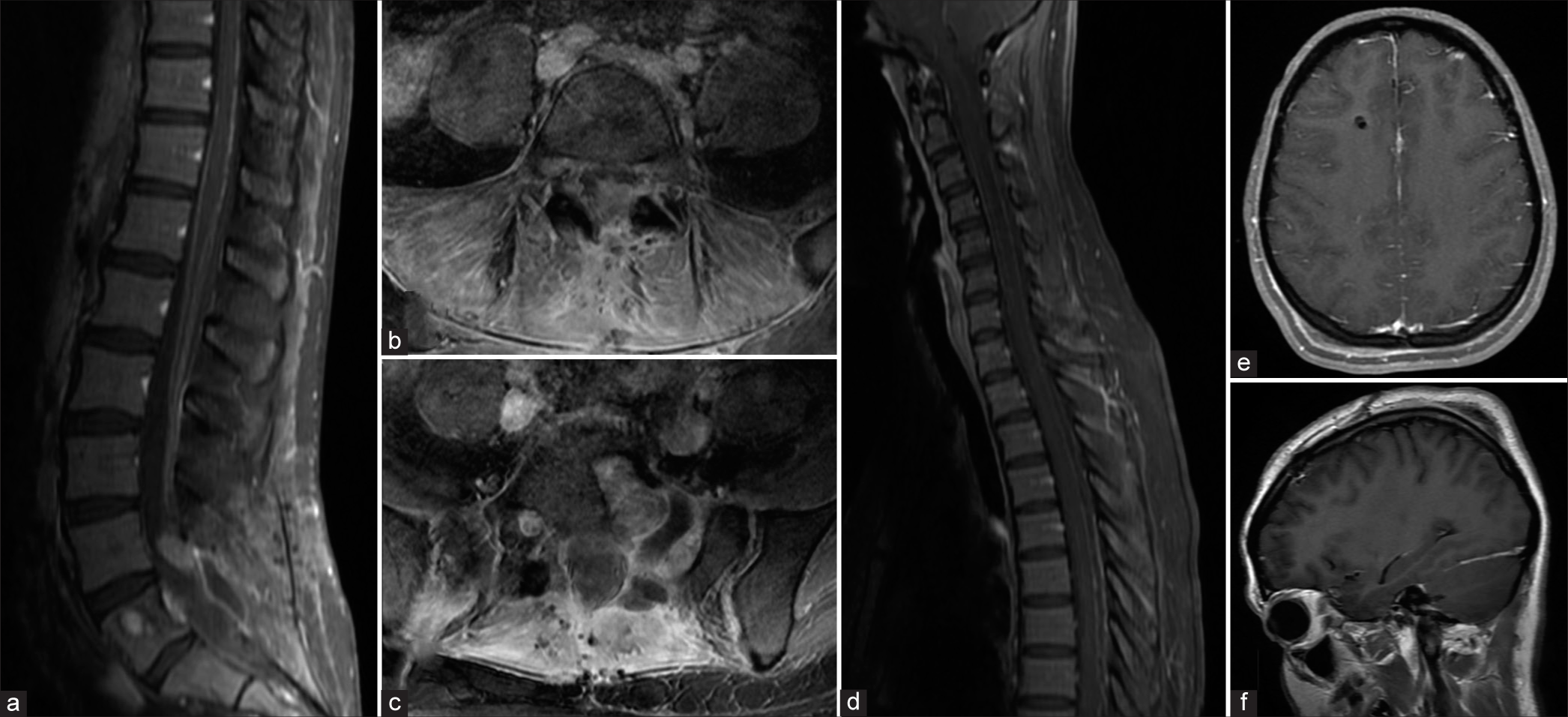
Figure 2:
MR images of the brain and whole spine demonstrating local disease recurrence and diffuse supratentorial and spinal leptomeningeal enhancement. (a) Sagittal T1-weighted MR with gadolinium contrast image of the lumbosacral spine demonstrating enhancing focus at L4/L5. (b) Axial T1-weighted MR image showing enhancing focus. (c) Axial T1-weighted MR image showing tumor cavity. (d) Sagittal T1-weighted MR image with gadolinium contrast demonstrating diffuse leptomeningeal enhancement along the cervical and thoracic spinal cord. (e) Axial T1-weighted MR image with gadolinium contrast demonstrating dural enhancement along the anterior aspect of the falx cerebri. (f) Sagittal T1-weighted MR image with gadolinium contrast demonstrating possible dural enhancement along the right tentorium.
A CT-guided lumbar puncture revealed only gelatinous material and core biopsy samples were taken. CSF studies showed high protein, low glucose, but no organisms. A frontal external ventricular drain was placed, and later converted to a ventriculoperitoneal shunt Craniospinal irradiation was administered after EVD insertion. Notably, the patient was discharged wheelchair-bound 3 months later.
Histology
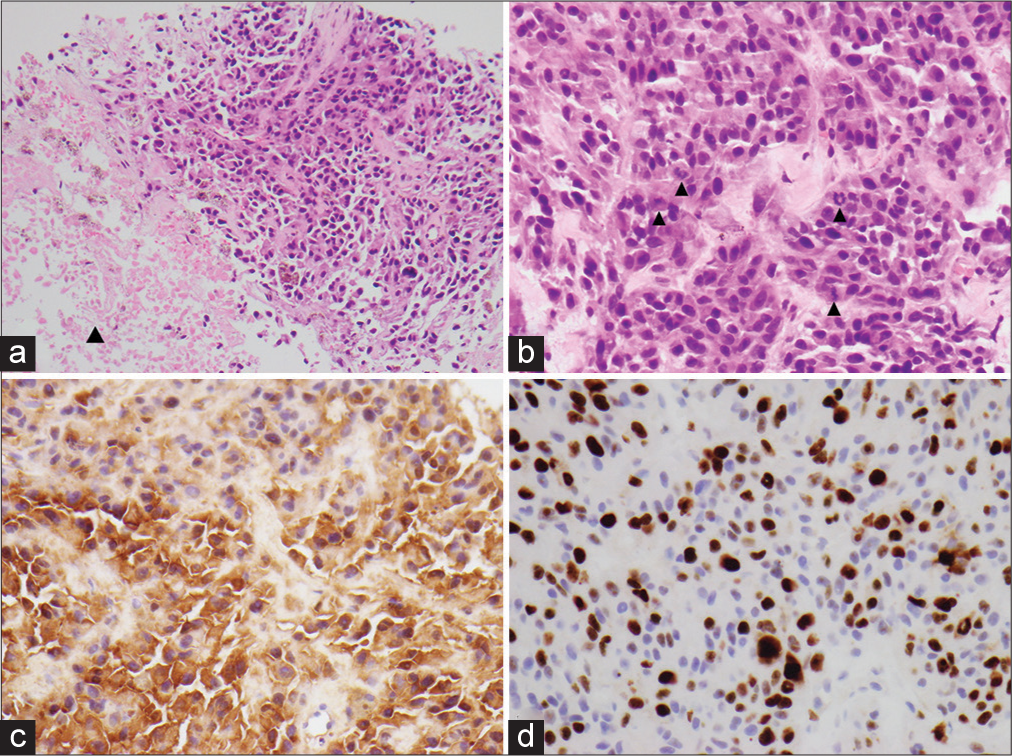
Tumor sections from both treatment periods showed a pigmented epithelioid to spindled cell tumor composed of cellular nodules, trabeculae and fascicles characterized by moderate nuclear pleomorphism, variably prominent nucleoli, and a moderate amount of eosinophilic cytoplasm [
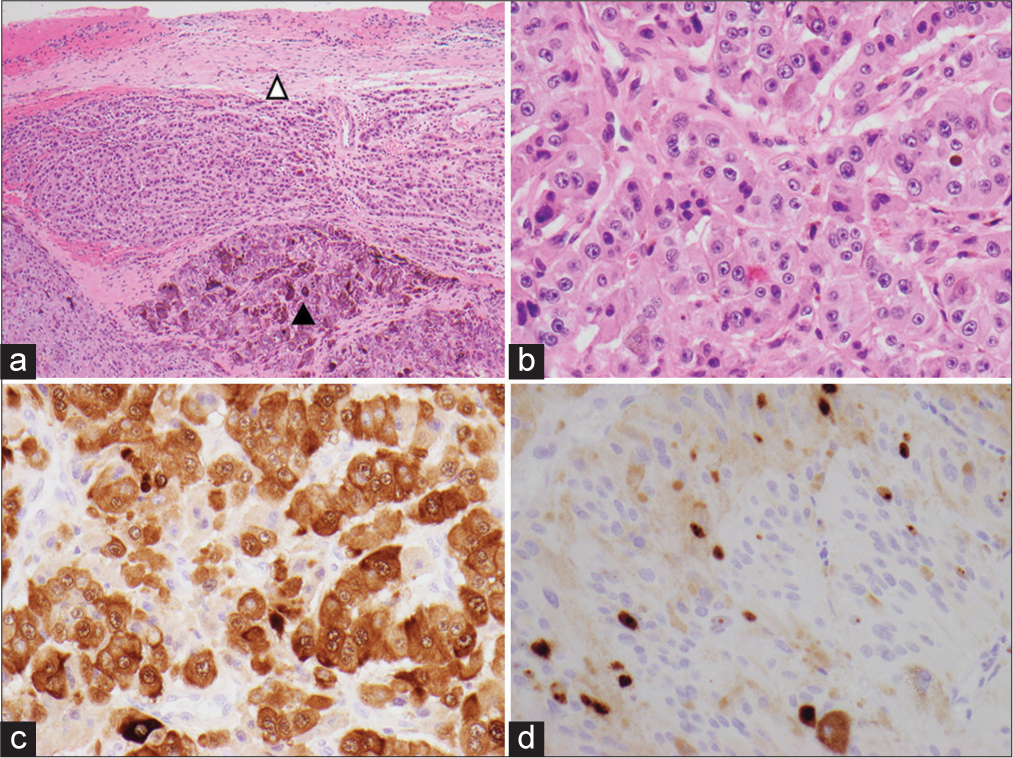
Figure 3:
Photomicrograph of the specimen. (a) Pigmented epithelioid to spindled cell tumor surrounded by fibrous rim suggestive of dura (open arrowhead. Closed arrowhead demonstrating more heavily pigmented tumor) (H&E, ×40). (b) Moderate nuclear pleomorphism and variably prominent nuclei (H&E, ×400). (c) S100 immunostain with brown chromogen, nonpigmented area of tumor (×400). (d) Ki67% immunostain, brown chromogen (×400).
DISCUSSION AND LITERATURE REVIEW
MMNSTs are rare aggressive tumors of presumed neural crest origin which most commonly involve the dorsal spinal nerves but may affect the spinal cord, sympathetic chain, cranial nerves, the lumbar plexus, peripheral nerves, and the gastrointestinal tract. Approximately 200 cases have been reported in the literature since the first identified case published in 1931 by Millar, who described the lesion as a “malignant melanotic tumor of the ganglion cells,”[
These tumors were reclassified in 2020 by the WHO as “MMNSTs.”[
Diagnostic study of choice
MR imaging remains the imaging modality of choice for these tumors. The lesions typically appear hyperintense on T1-weighted images and hypointense on T2-weighted images.[
Treatment recommendations
At present, there is no optimal treatment protocol for managing MMNSTs. Most studies advocate maximal safe resection. Radiotherapy is often offered only following local recurrence or distal metastases (i.e., for palliative symptom control). In this case, our patient did not originally receive adjuvant radiotherapy, as there was no evidence of residual tumor (i.e., following gross total resection). In this case and for patients with recurrent tumors or evidence of holospinal/brain leptomeningeal spread, craniospinal radiation with focal RT boosts to local lesions may be utilized.
CONCLUSION
MMNSTs are rare tumors that have a high propensity for local recurrence or metastasizing through the leptomeninges. Treatment recommendations include gross total initial resection followed by adjuvant radiotherapy and/or chemoimmunotherapy.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Alexiev BA, Chou PM, Jennings LJ. Pathology of melanotic schwannoma. Arch Pathol Lab Med. 2018. 142: 1517-23
2. Carney JA. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol. 1990. 4: 206-22
3. Kallen ME, Hornick JL. The 2020 WHO classification: What’s new in soft tissue tumor pathology?. Am J Surg Pathol. 2021. 45: e1-23
4. Millar WG. A malignant melanotic tumour of ganglion cells arising from a thoracic sympathetic ganglion. J Pathol Bacteriol. 1932. 35: 351-7
5. Solomou G, Dulanka Silva AH, Wong A, Pohl U, Tzerakis N. Extramedullary malignant melanotic schwannoma of the spine: Case report and an up to date systematic review of the literature. Ann Med Surg (Lond). 2020. 59: 217-23
6. Torres-Mora J, Dry S, Li X, Binder S, Amin M, Folpe AL. Malignant melanotic schwannian tumor: A clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am J Surg Pathol. 2014. 38: 94-105

