

- Department of Neurosurgery, University Hospital Center Zagreb, Zagreb, Croatia.
- Department of Pediatric Infectious Diseases, University Hospital for Infectious Diseases, Dr. Fran Mihaljevic, Zagreb, Croatia.
- Department of Pediatrics, Sestre Milosrdnice University Hospital Center, Zagreb, Croatia.
- Department of Pathology, Clinical Medical Centre Zagreb, Zagreb, Croatia.
- Department of Pediatrics, Division of Hematology and Oncology, Children’s Hospital Zagreb, Zagreb, Croatia.
Correspondence Address:
Tena Trbojevic, Department of Pediatrics, Sestre milosrdnice University Hospital Center, Zagreb, Croatia.
DOI:10.25259/SNI_867_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Jakob Nemir1, Lorna Stemberger Maric2, Tena Trbojevic3, Kamelija Zarkovic4, Filip Jadrijević-Cvrlje5. Papillary tumor of the pineal region in pediatric patient – A case report. 28-Oct-2022;13:488
How to cite this URL: Jakob Nemir1, Lorna Stemberger Maric2, Tena Trbojevic3, Kamelija Zarkovic4, Filip Jadrijević-Cvrlje5. Papillary tumor of the pineal region in pediatric patient – A case report. 28-Oct-2022;13:488. Available from: https://surgicalneurologyint.com/surgicalint-articles/11967/
Date of Submission
18-Sep-2022
Date of Acceptance
24-Sep-2022
Date of Web Publication
28-Oct-2022
Abstract
Background: Papillary tumor of the pineal region (PTPR) represents a rare and histologically distinct subgroup of tumors originating in the pineal region. Few pediatric cases have been reported so far in the literature; therefore, clinical data are scarce.
Case Description: We describe a case of PTPR in a 9-year-old girl who presented with a 5-month history of excessive appetite and weight gain. The patient underwent neuroimaging procedures and total gross surgical resection with postoperative adjuvant local radiotherapy, which from our experience was the best treatment choice as an attempt to avoid local recurrence. During 78-month follow-up, the patient from our study manifested no disease recurrence.
Conclusion: PTPR should be included in the differential diagnosis of pineal region masses.
Keywords: Central nervous system, Children, Immunohistochemistry, Papillary tumor of the pineal region, Pineal region
INTRODUCTION
Tumors of the pineal region account for about 3–11% of all brain tumors in childhood. Papillary tumor of the pineal region (PTPR) is a rare neuroepithelial tumor originating in this area.[
PTPR affects mostly young adults in the third decade of life.[
CASE DESCRIPTION
A 9-year-old girl presented with a 5-month history of excessive appetite and weight gain. Patient’s mother noticed 2–3 weeks before admission, an adjustment of patient’s head during specific activities (i.e., watching television). One week before admission, the patient suffered from mild frontal headache, and manifested blurred vision as well as broad-based gait. On the day before admission to hospital, diplopia occurred. Physical and neurological examination was unremarkable except for unstable gait and diplopia in all directions. Initially bilateral papilledema was found; therefore, neuroimaging was performed. Brain magnetic resonance imaging (MRI) revealed a triventricular hypertensive hydrocephalus due to heterogeneously-enhanced and space-occupying lesion 3.3 × 3 cm in diameter with limited cystic and hemorrhagic components in the pineal region. The tumor was localized; extended temporarily from the roof of the fourth ventricle (filling the cerebral aqueduct) with infiltration of the mesencephalon tectum and separation of the cerebral peduncles. Diffusion restriction and T2-hyperintense signal indicated tumor hypercellularity [
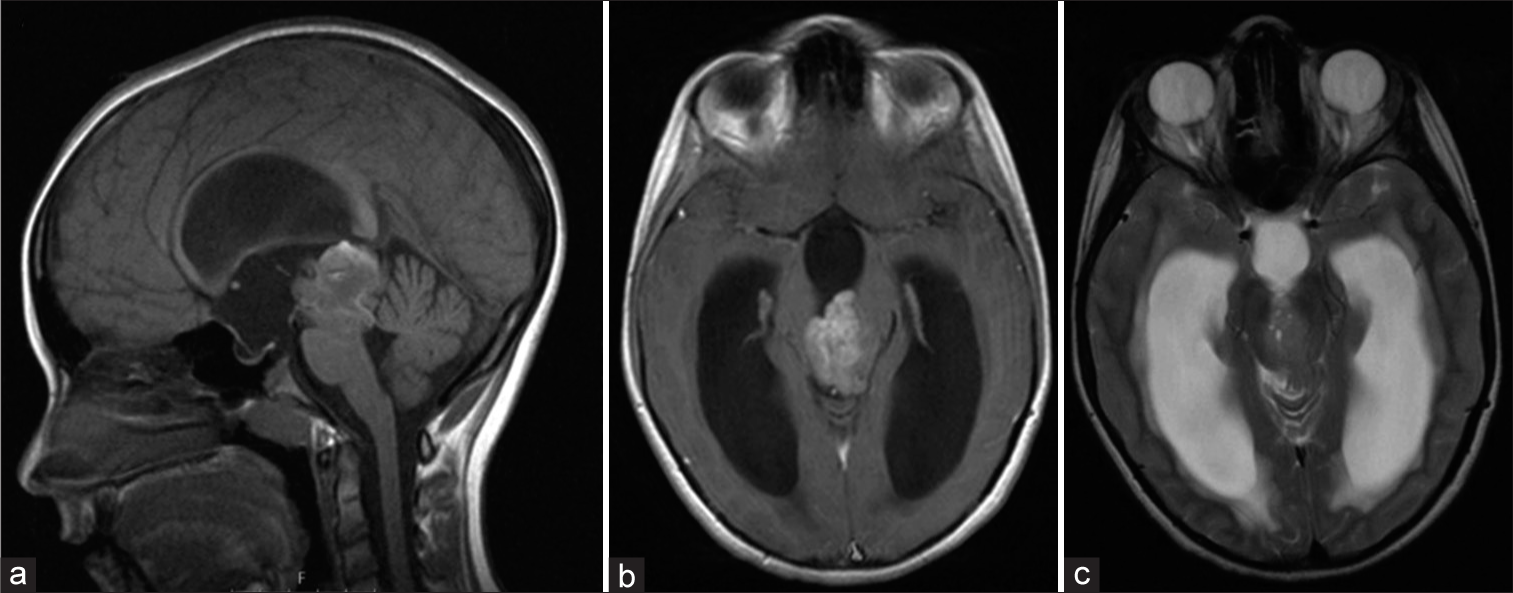
Figure 1:
Preoperative magnetic resonance imaging (MRI). T1 contrast-enhanced midsagittal MRI displays a tumor mass located in the pineal region, demonstrating heterogeneous post contrast opacification, signs of hemorrhage, and cystic areas (a and b). T2-weighted axial MRI reveals tumor hypercellularity, a ballooned third ventricle and enlarged lateral ventricles-signs of hydrocephalus (c).
The initial treatment option was to perform an endoscopic third ventriculostomy to treat hydrocephalus and to simultaneously obtain a tumor specimen for histopathological analysis, which revealed a PTPR. A median suboccipital craniotomy was preformed 14 days later; the pineal region was reached through a supracerebellar-infratentorial approach, followed by complete resection of the tumor.
The definitive histopathological analysis revealed tumor tissue composed of papillary formations and cellular tumor with ependymal-like differentiation. Eosinophilic columnar or elongated cells were present in papillary areas around the vessels, while in ependymal-like areas gliovascular rosettes and tubes were noticed. The nuclei of tumor cells were round to oval, while atypical nuclei were only occasionally present. Mitotic activity ranges from 4 to 10/10 HPF, while Ki67 values range up to 8%. Microvascular proliferation was not present. Large foci of necrosis were without pseudopalisades of the surrounding tumor cells. Tumor tissue was diffusely immunopositive for AE1/AE3, CK18, neuron-specific enolase (NSE), S100, and vimentin, but was only sporadically positive for synaptophysin. Nuclear INI1 expression was present in tumor cells. Neu-N was not seen. Neurofilament protein was evaluated with the aim of exclusion of pineocytomas, and it was negative. Chromogranin as marker of pinealoblastoma (PB) was negative. Epithelial membrane antigen (EMA) and glial fibrillary acidic protein (GFAP) immunostain were negative. Therefore, histological diagnosis of the tumor was PTPR [
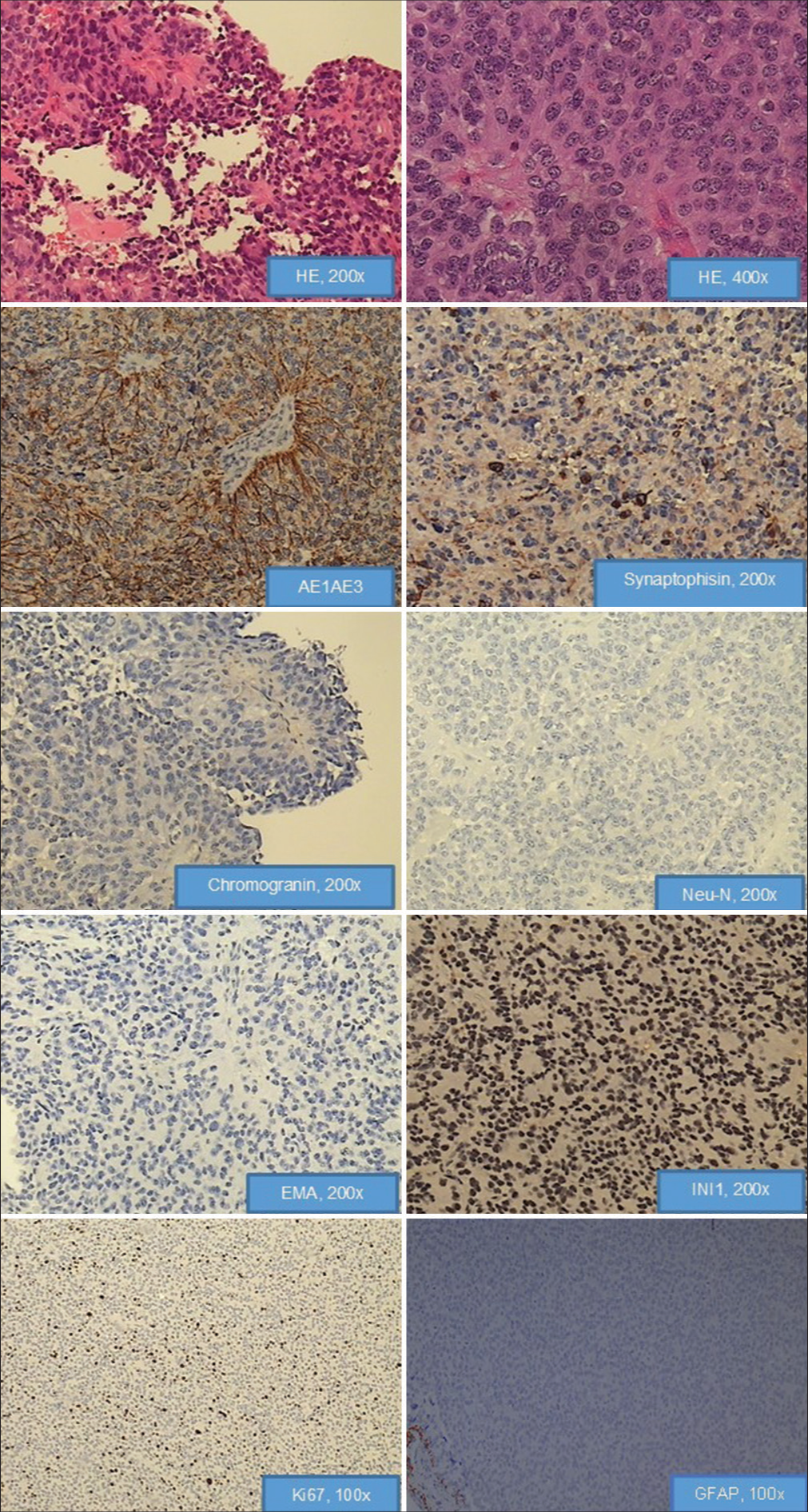
Figure 2:
Hematoxylin and eosin-stained sections of paraffin embedded tissue show tumor tissue composed of papillary formations and cellular tumor with ependymal-like differentiation. Eosinophilic columnar or elongated cells are present in papillary areas around the vessels, while in ependymal-like areas gliovascular rosettes and tubes are noticed. The nuclei of tumor cells are round to oval, while atypical nuclei are only occasionally present. Microvascular proliferation is not present. Large foci of necrosis are without pseudopalisades of the surrounding tumor cells. Tumor tissue is diffusely immunopositive for AE1/AE3, sporadically positive for synaptophysin neuron-specific enolase, negative for chromogranin, Neu-N, and Epithelial membrane antigen. Nuclear INI1 expression is present in tumor cells. Mitotic activity ranges from 4 to 10 per 10 HPF, while Ki67 values range from 2% to 8%. Tumor showed complete absence of immunoreactivity for glial fibrillary acidic protein.
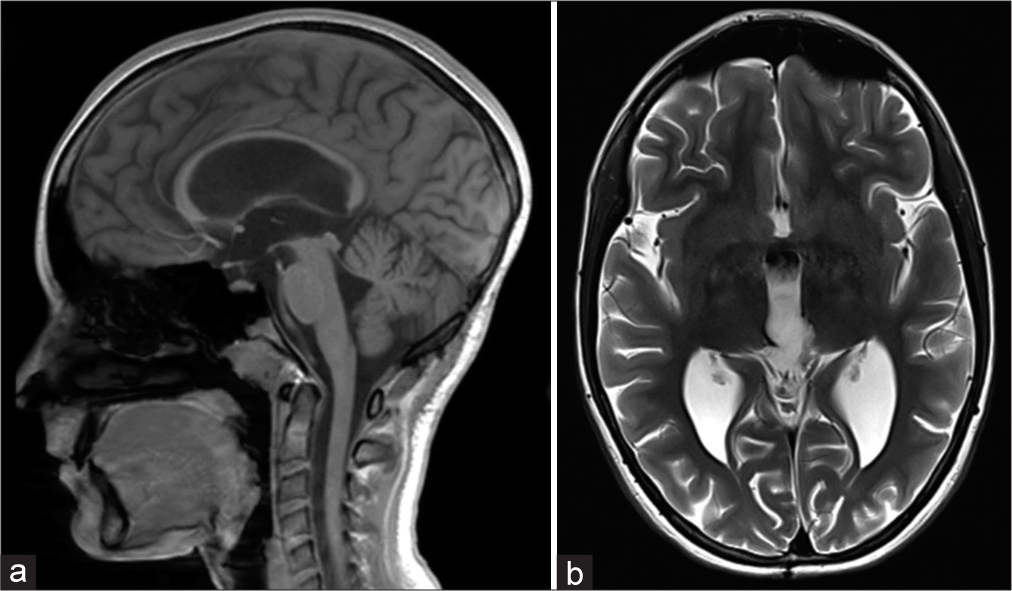
Postoperative, vertical supranuclear gaze palsy, convergence deficit, and dilated pupils (Parinaud’s syndrome) were noticed what led to mild gait instability. The patient also had need for cortisol supplementation. Postoperative MRI of brain and total spinal cord found no residual tumor or signs of leptomeningeal dissemination [
DISCUSSION
Primary tumors of the pineal region with papillary features are very rare.[
CONSCLUSION
The PTPR is a rare tumor that must be included in the differential diagnosis of pineal region masses. Regarding treatment options, as we have demonstrated in our case study, local radiotherapy could be provided after gross total resection with the aim to avoid local recurrence. However, long-term follow-up is required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Abela L, Rushing EJ, Ares C, Scheer I, Bozinov O, Boltshauser E. Pediatric papillary tumors of the pineal region: To observe or to treat following gross total resection?. Childs Nerv Syst. 2013. 29: 307-10
2. Aggarwal SK, Agarwal P, Sahu RN. Papillary tumor of pineal region with an unusual clinical presentation: Case report and review of the literature. Asian J Neurosurg. 2016. 11: 78-9
3. Biswas R, Kumari K, Haresh KP, Gupta S, Halder A, Rath GK. Papillary tumor of the pineal region in an adolescent. Indian J Med Paediatr Oncol. 2019. 40: 199-201
4. Boßelmann CM, Gepfner-Tuma I, Schittenhelm J, Brendle C, Honegger J, Tabatabai G. Papillary tumor of the pineal region: A single-center experience. Neurooncol Pract. 2020. 7: 384-90
5. Buffenoir K, Rigoard P, Wager M, Ferrand S, Coulon A, Blanc JL. Papillary tumor of the pineal region in a child: Case report and review of the literature. Childs Nerv Syst. 2008. 24: 379-84
6. Choque-Velasquez J, Colasanti R, Resendiz-Nieves J, Jahromi BR, Tynninen O, Collan J. Papillary tumor of the pineal region in children: Presentation of a case and comprehensive literature review. World Neurosurg. 2018. 117: 144-52
7. Cimino PJ, Gonzalez-Cuyar LF, Perry A, Dahiya S. Lack of BRAF-V600E mutation in papillary tumor of the pineal region. Neurosurgery. 2015. 77: 621-8
8. Dagnew E, Langford LA, Lang FF, DeMonte F. Papillary tumors of the pineal region: Case report. Neurosurgery. 2007. 60: E953-5
9. Drummond KJ, Rosenfeld JV. Pineal region tumours in childhood. A 30-year experience. Childs Nerv Syst. 1999. 15: 119-26
10. Fauchon F, Hasselblatt M, Jouvet A, Champier J, Popovic M, Kirollos R. Role of surgery, radiotherapy and chemotherapy in papillary tumors of the pineal region: A multicenter study. J Neurooncol. 2013. 112: 223-31
11. Favero G, Bonomini F, Rezzani R. Pineal gland tumors: A review. Cancers (Basel). 2021. 13: 1547
12. Fèvre-Montange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, Champier J, Saint-Pierre G. Prognosis and histopathologic features in papillary tumors of the pineal region: A retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006. 65: 1004-11
13. Fèvre Montange M, Vasiljevic A, Fouquet AM, Bernier M, Champier J, Chrétien F. Histopathologic and ultrastructural features and claudin expression in papillary tumors of the pineal region: A multicenter analysis. Am J Surg Pathol. 2012. 36: 916-28
14. Fèvre Montange M, Vasiljevic A, Champier J, Jouvet A. Papillary tumor of the pineal region: Histopathological characterization and review of the literature. Neurochirurgie. 2015. 61: 138-42
15. Gupta K, Khursheed S, Nayil K, Khursheed S, Makhdoomi R. Papillary tumor of pineal region in a 5-year-old male child: A rare entity. Asian J Neurosurg. 2021. 16: 824-6
16. Gutenberg A, Brandis A, Hong B, Gunawan B, Enders C, Schaefer IM. Common molecular cytogenetic pathway in papillary tumors of the pineal region (PTPR). Brain Pathol. 2011. 21: 672-7
17. Hasselblatt M, Blumcke I, Jeibmann A, Rickert CH, Jouvet A, Van de Nes JA. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Neuropathol Appl Neurobiol. 2006. 32: 278-83
18. Heim S, Beschorner R, Mittelbronn M, Keyvani K, Riemenschneider MJ, Vajtai I. Increased mitotic and proliferative activity are associated with worse prognosis in papillary tumors of the pineal region. Am J Surg Pathol. 2014. 38: 106-10
19. Hua X, Yang P, Zhang M, Zhao Y, Wang B. Papillary tumor of the pineal region: A case report and review of the literature. Exp Ther Med. 2015. 10: 1375-79
20. Iacoangeli M, Colasanti R, Esposito D, Di Rienzo A, Di Somma L, Dobran M. Supraorbital subfrontal trans-laminar endoscope-assisted approach for tumors of the posterior third ventricle. Acta Neurochir (Wien). 2017. 159: 645-54
21. Jouvet A, Fauchon F, Liberski P, Saint-Pierre G, Didier-Bazes M, Heitzmann A. Papillary tumor of the pineal region. Am J Surg Pathol. 2003. 27: 505-12
22. Junior GV, Dellaretti M, De Carvalho GT, Brandão RA, Mafra A, De Sousa AA. Papillary tumor of the pineal region. Brain Tumor Patholo. 2011. 28: 329-34
23. Kaloshi G, Rroji A, Lame A, Leka L, Haxhihyseni E, Vreto G. Natural history of papillary tumor of the pineal region: New insights on biological explanation. J Neurooncol. 2010. 100: 487-8
24. Kawahara I, Tokunaga Y, Yagi N, Iseki M, Abe K, Hayashi T. Papillary tumor of the pineal region. Neurol Med Chir (Tokyo). 2007. 47: 568-71
25. Li J, Recinos PF, Orr BA, Burger PC, Jallo GI, Recinos VR. Papillary tumor of the pineal region in a 15-month-old boy. J Neurosurg Pediatr. 2011. 7: 534-8
26. Lombardi G, Poliani PL, Manara R, Berhouma M, Minniti G, Tabouret E. Diagnosis and treatment of pineal region tumors in adults: A EURACAN overview. Cancers (Basel). 2022. 14: 3646
27. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007. 114: 97-109
28. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, FigarellaBranger D. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021. 23: 1231-51
29. Marcol W, Kotulska K, Grajkowska W, Gołka D, Właszczuk P, Drogosiewicz M. Papillary pineocytoma in child: A case report. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2007. 151: 121-3
30. Mathkour M, Hanna J, Ibrahim N, Scullen T, Kilgore MD, Werner C. Papillary tumor of the pineal region in pediatric populations: An additional case and systematic review of a rare tumor entity. Clin Neurol Neurosurg. 2021. 201: 106404
31. Mehta N, Gupta G, Shaikh S. Recurrent papillary tumor of pineal region misdiagnosed as pineocytoma 9-years ago. Asian J Neurosurg. 2021. 16: 398-401
32. Nakamura H, Makino K, Kochi M, Nakazato Y, Kuratsu J. Successful treatment of neoadjuvant therapy for papillary tumor of the pineal region. Brain Tumor Pathol. 2009. 26: 73-7
33. Narolahi H, Geramizadeh B, Ansari M, Hamedi SH. Papillary tumor of the pineal region: A case report. Middle East J Cancer. 2018. 9: 165-9
34. Patil M, Karandikar M. Papillary tumor of pineal region: A rare entity. Asian J Neurosurg. 2016. 11: 453
35. Poulgrain K, Gurgo R, Winter C, Ong B, Lau Q. Papillary tumour of the pineal region. J Clin Neurosci. 2011. 18: 1007-17
36. Rickard KA, Parker JR, Vitaz TW, Plaga AR, Wagner S, Parker JC. Papillary tumor of the pineal region: Two case studies and a review of the literature. Ann Clin Lab Sci. 2011. 41: 174-81
37. Sato TS, Kirby PA, Buatti JM, Moritani T. Papillary tumor of the pineal region: Report of a rapidly progressive tumor with possible multicentric origin. Pediatr Radiol. 2009. 39: 188-90
38. Vaghela V, Radhakrishnan N, Radhakrishnan VV, Menon G, Kesavadas C, Thomas B. Advanced magnetic resonance imaging with histopathological correlation in papillary tumor of pineal region: Report of a case and review of literature. Neurol India. 2010. 58: 928-32
39. Yamaki VN, Solla DJ, Ribeiro RR, Da Silva SA, Teixeira MJ, Figueiredo EG. Papillary tumor of the pineal region: Systematic review and analysis of prognostic factors. Neurosurgery. 2019. 85: E420-9
40. Yano H, Ohe N, Nakayama N, Shinoda J, Iwama T. Clinicopathological features from long-term observation of a papillary tumor of the pineal region (PTPR): A case report. Brain Tumor Pathol. 2009. 26: 83-8

