

- Department of Neurobiology, Drexel University College of Medicine, Philadelphia, USA
- Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA
- Department of Radiology, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA
Correspondence Address:
Sandi Lam
Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA
DOI:10.4103/2152-7806.196921
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Michael G. Z. Ghali, Visish M. Srinivasan, Arvind C. Mohan, Jeremy Y. Jones, Peter T. Kan, Sandi Lam. Pediatric cerebral cavernous malformations: Genetics, pathogenesis, and management. 28-Dec-2016;7:
How to cite this URL: Michael G. Z. Ghali, Visish M. Srinivasan, Arvind C. Mohan, Jeremy Y. Jones, Peter T. Kan, Sandi Lam. Pediatric cerebral cavernous malformations: Genetics, pathogenesis, and management. 28-Dec-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/pediatric-cerebral-cavernous-malformations-genetics-pathogenesis-and-management/
Date of Submission
06-Apr-2016
Date of Acceptance
14-Jun-2016
Date of Web Publication
28-Dec-2016
Keywords: Cerebral cavernous malformation, cavernous angioma, cavernous hemangioma, vascular lesion, neurovascular, intracranial
CASE REPORTS
Case 1
A 2-year-old Hispanic girl presented with a 3-day history of vomiting followed by a sudden onset of right facial droop and right eye ptosis. She had achieved normal developmental milestones until then. In retrospect, she had several months of occasional left leg weakness causing falls. There was no history of seizures. She had a family history with two second-degree relatives with cerebral cavernous malformations (CCMs) of the cerebrum and brainstem. She had not received prior imaging or screening. Genetic testing revealed a heterozygous mutation in the KRIT1 gene.
Computerized tomography (CT) of the head demonstrated multiple hemorrhagic lesions of different ages, with the largest being a 3 × 2.5 cm lesion in the right thalamus with mesencephalic extension, along with 1 cm lesions in the left forceps minor and right atrium. There was obstructive hydrocephalus without intraventricular hemorrhage [
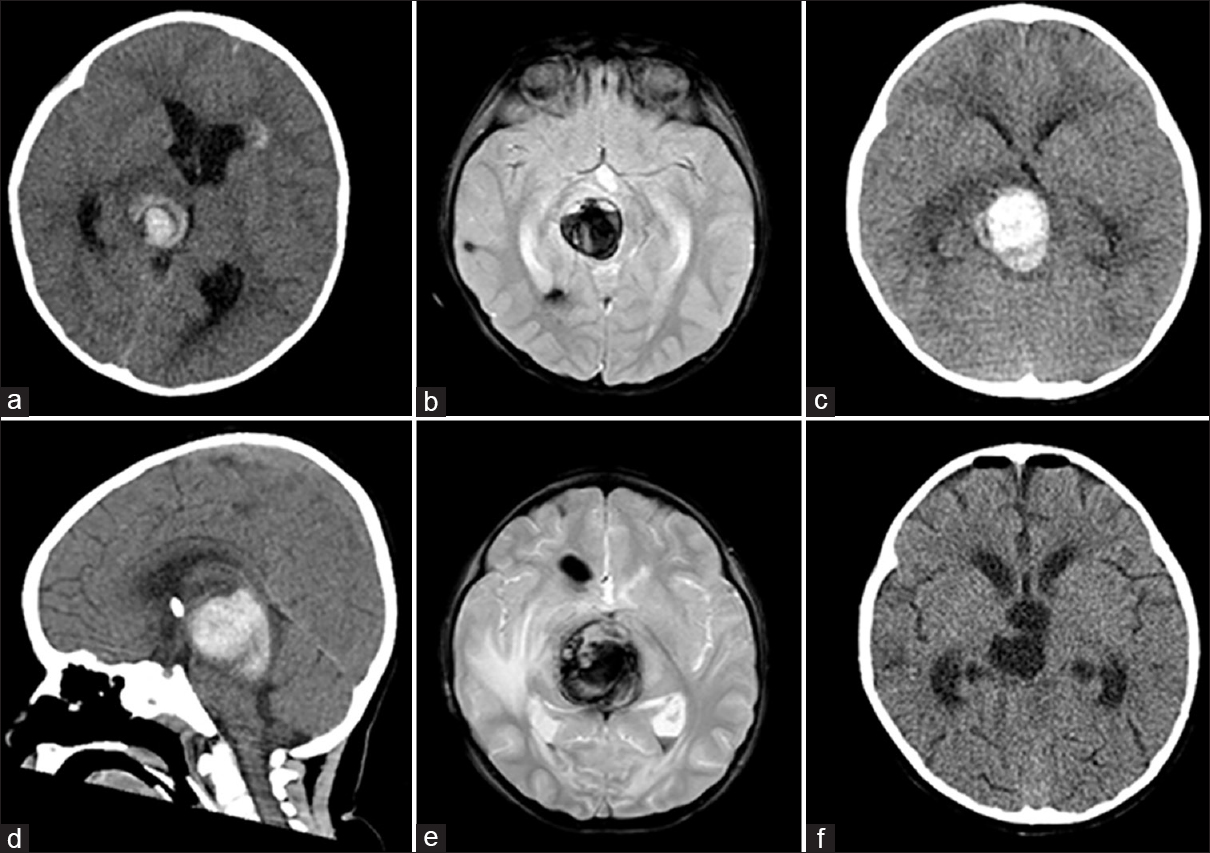
Figure 1
Evolution of hemorrhage from familial cavernous malformation (Case 1). (a) Computerized tomography (CT) of head at presentation, showing 2.5 × 3 cm hemorrhagic CM in right thalamic-mesencephalic junction. (b) Magnetic resonance imaging (MRI) of the brain, axial gradient echo sequence showing the same. (c) Expansion of hemorrhage to 3.5 × 3 × 3 cm. (d) CT head, sagittal section, day 9, showing further extension in the craniocaudal dimension. (e) Preoperative MRI brain, axial gradient echo sequence, showing lesion growing to 3.3 cm anteroposterior × 3.7 cm transverse × 3.7 cm craniocaudal. (f) Postoperative CT head showing complete resection of lesion and hematoma
An external ventricular drain (EVD) was placed. Magnetic resonance imaging (MRI) of the brain characterized these lesions as CCMs [
Case 2
A 7-year-old girl presented with diplopia, ataxia, and headaches. Imaging revealed a hemorrhagic right middle cerebellar peduncle lesion suggestive of a solitary CCM [
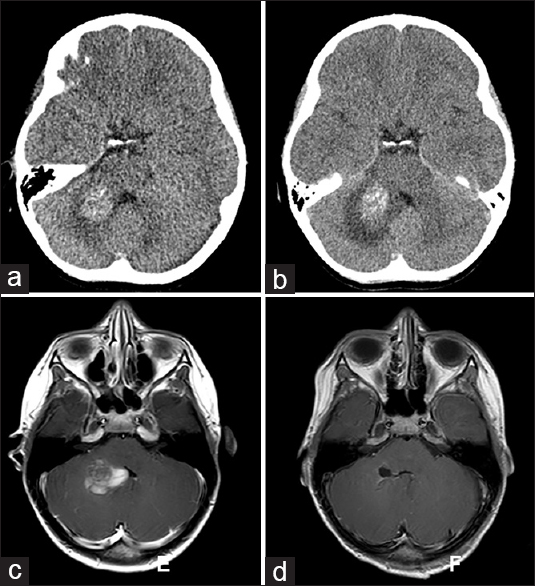
Figure 2
Evolution of hemorrhage in brainstem cavernous malformation (Case 2). (a) Computerized tomography (CT) of the head at presentation showing hemorrhage in right middle cerebellar peduncle that does not extend to the pial surface. (b) CT head 1 month later during recurrence of symptoms shows worsened perilesional edema and progression toward the ependymal surface of the fourth ventricle. (c) Magnetic resonance imaging (MRI) of the brain T1-weighted sequence with contrast at the time of second hemorrhage demonstrates surgical corridor made possible by hemorrhage extending to the right lateral recess of fourth ventricle. (d) Postoperative MRI demonstrates complete resection of lesion
REVIEW
Epidemiology
The epidemiology of CCMs, or cavernomas, is an area of extensive investigation in adults. However, less data exist regarding their incidence and prevalence in the pediatric age group. Moreover, a prior false assumption of an exclusively congenital etiology may have led to an underestimation of the overall hemorrhage risk. The development of CCMs appears to increase with age, reaching a plateau in late adolescence, as demonstrated by Al-Holou et al. in 2012.[
The overall incidence for the development of new CCMs in children is correlated with their pre-existing cavernoma burden. Gross et al. reported incidences of approximately 1.2% per patient per year and 2.5% per lesion per year; that is, the presence of multiple CMs confers a greater risk of de novo cavernoma-genesis compared with patients with solitary CMs (7.1% versus 0.6% per lesion per year).[
Histopathology
Cavernous malformations (CMs) are hamartomatous, cystically-dilated vascular spaces composed of a single layer of endothelium with possible infrequent subendothelial cells, without elastic lamina or smooth muscle cells, embedded in a collagenous extracellular matrix.[
Clinical presentation and diagnosis
Approximately 85% of CMs are supratentorial in children (92% lobar, 8% deep). The remainder are located infratentorially (57% brainstem, 43% cerebellar), with rare occurrence in the spinal cord.[
By definition, all CCMs exhibit varying degrees of microhemorrhage, as evidenced by hemosiderin deposition. When located in noneloquent areas of the brain, slightly larger degrees of hemorrhage may be tolerated. Concern arises when CCMs occur in critical brain regions, such as the brainstem, where even small hemorrhages may compromise vital functions.
The overall risk for CCM hemorrhage in the pediatric population is approximately 0.5% per lesion per year.[
Imaging
CT scanning has poor sensitivity for detection of CCMs. T2-weighted MRI, especially gradient echo (GRE) or susceptibility weighted imaging (SWI) sequences, possesses the greatest sensitivity for detection of CCMs and reveals a mixed signal core and surrounding low signal rim, often with evidence of microhemorrhages. Brain MRI screening is indicated for first-degree relatives of patients with CMs with two or more affected family members.
Developmental venous anomalies (DVAs) are seen in up to 20% of patients with CMs,[
Genetics and etiopathogenesis
CCMs may occur sporadically or be transmitted in an autosomal dominant fashion with variable penetrance; the familial type has been reported to account for roughly half of the cases in Hispanics and up to one-fifth of the cases in Caucasians, and is also associated with a greater annual risk of symptomatic bleeding.[
Three genetic loci have been associated with and account for ~80% of familial CCM: CCM1/Krit1, CCM2/MGC4607, and CCM3/PDCD10.[
Furthermore, single nucleotide polymorphisms (SNPs) of inflammatory and immune response genes have been associated with different features of CCM natural history, including disease burden and severity of risk of intracerebral hemorrhage; identified SNPs include IL-1RN, TGFBR2, CHUK, SELS, CD3G, IGH, and IGL. This information may have implications for risk stratification and treatment planning.
A Knudsonian two-hit hypothesis has been presented to account for all cases of CCM. This appears to hold true for the familial form, wherein an inherited germline CCM mutation and a single acquired somatic mutation in the homologue affects cavernoma-genesis,[
Molecular pathogenesis
The molecular pathogenesis of CCMs is linked to the gene products of CCM1, CCM2, and CCM3, which are also known as Krev Interaction Trapped 1 (Krit1), malcavernin, and PDCD10, respectively.

CCM1 encodes Krit1, a microtubule-associated protein that also interacts with Rap1A, ICAP-1, and a variety of other proteins. Rap1A is a Ras-family GTPase involved in cellular differentiation and morphogenesis, as well as regulation of cellular polarity and cytoskeletal organization.[
In addition, Krit1 may contribute to regulation of transmembrane β1-integrin-mediated signal transduction and cell-cell as well as cell-extracellular matrix (ECM) signaling, both of which are critical in the formation of microtubules and, in turn, endothelial structure/function and angiogenesis. An important intermediary protein in β1-integrin-mediated signal transduction is ICAP-1, which binds the cytoplasmic domain and links it to the cytoskeleton. Further, ICAP possesses binding sites for Krit1, which may play a regulatory function in the β1-integrin/ICAP-1 interaction.[
Krit1 also interacts with malcavernin, a scaffold protein that associates with mitogen-activated protein kinase (MAPK)-extracellular-regulated kinase (ERK) kinase 3 (MEKK3), a pathway critical in the regulation of endothelial proliferation and migration, adhesion, and cytoskeletal regulation.[
Programmed cell death 10 gene (PDCD10) encodes for a protein involved in apoptosis and associates with both Krit1 and malcavernin.[
CCM1-3 share their ability to negatively modulate RhoA/Rho kinase;[
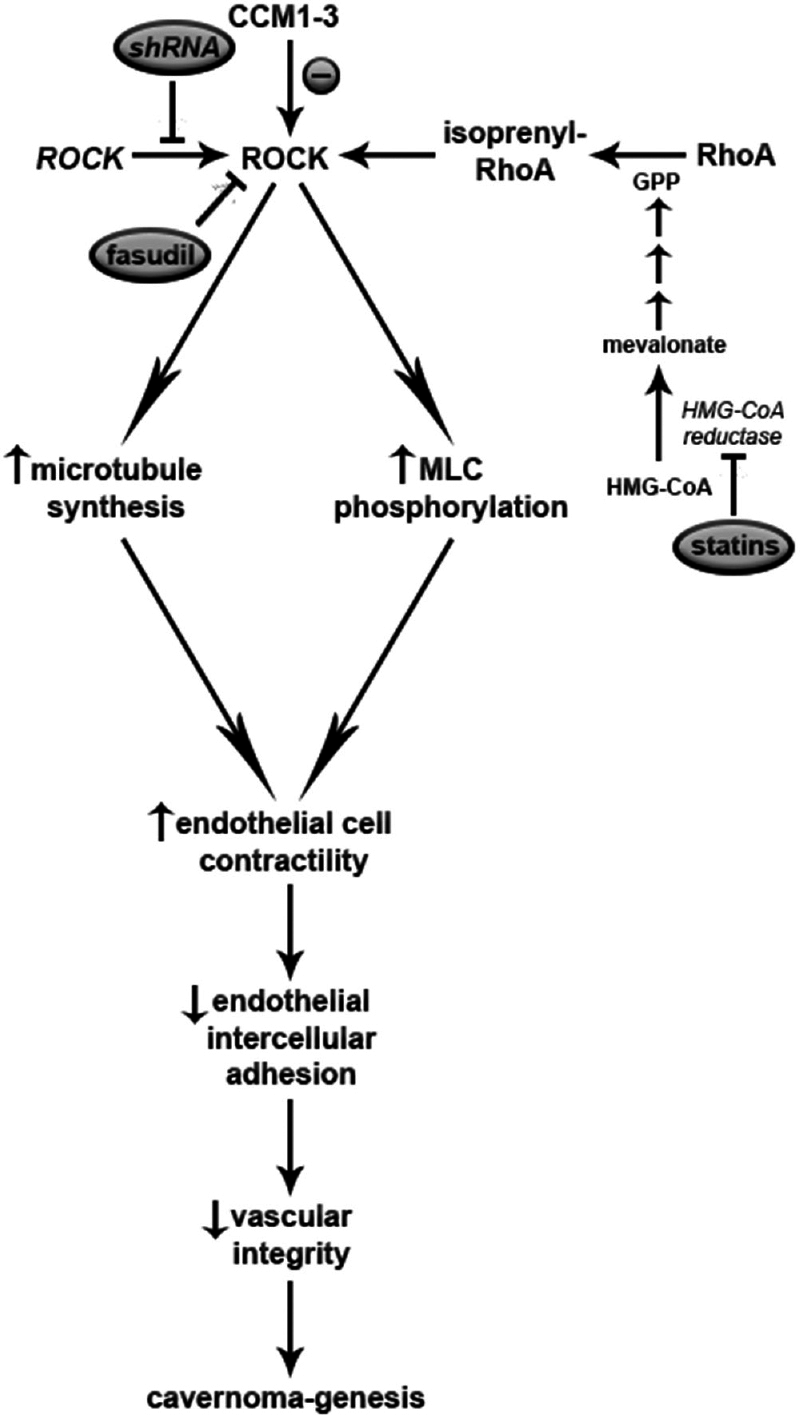
Figure 3
Summary of main pathways of cavernoma-genesis and potential targeted therapies. The molecular pathogenesis of familial cavernomas centers around modulation of Rho Kinase (ROCK), which modulates microtubule synthesis. This, in turn, alters contractility of endothelial cells, intercellular adhesion, and vascular integrity. Loss of vascular integrity allows cavernoma-genesis
Treatment
Standard management options for CCMs have classically included observation and surgical removal.[
Especially for deep-seated CCMs in the pons or brainstem, surgeons typically prefer to wait for the CCM lesion to present to a surgically accessible surface without the need for direct surgical dissection through eloquent tracts.[
Surgical resection of CMs in the setting of epilepsy is a topic of continuing investigation. In this scenario, the hemosiderin ring associated with CCMs is associated with epileptogenic potential[
Evolution of minimally invasive therapies
Stereotactic radiosurgery (SRS) has been reported by some groups as a potential treatment option.[
Magnetic resonance-guided focused ultrasound (MRgFUS) has been reported as a novel minimally invasive procedure for the treatment of central nervous system pathologies, including CCMs.[
Another minimally invasive treatment option for CCMs may include MR-guided laser interstitial thermal therapy (MRgLITT), also called MR-guided stereotactic laser ablation (MRgSLA). MRgSLA has been used in the treatment of tumors, epilepsy, and chronic pain syndromes. A non-negligible risk of focal neurologic deficits has been reported, especially for deep targets.[
CONCLUSION
CCMs are a common cause of intracranial hemorrhage in the pediatric population. A significant proportion of CCMs identified in pediatric patients, especially those with a history of symptomatic hemorrhage, may be associated with a familial subtype with identifiable genetic mutations in genes CCM1, CCM2, or CCM3. Future research will further identify genetic pathophysiology, risk of rupture, and risk of CCM formation based on genotyping. Surgery remains the gold standard of treatment. Directions for future evaluation include minimally invasive procedures, as well as potential for an increased role of medical management using targeted molecular therapies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abla AA, Turner JD, Mitha AP, Lekovic G, Spetzler RF. Surgical approaches to brainstem cavernous malformations. Neurosurg Focus. 2010. 29: E8-
2. Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 2009. 18: 919-30
3. Al-Holou WN, O’Lynnger TM, Pandey AS, Gemmete JJ, Thompson BG, Muraszko KM. Natural history and imaging prevalence of cavernous malformations in children and young adults. J Neurosurg Pediatr. 2012. 9: 198-205
4. Al-Shahi Salman R, Hall JM, Horne MA, Moultrie F, Josephson CB, Bhattacharya JJ. Untreated clinical course of cerebral cavernous malformations: A prospective, population-based cohort study. Lancet Neurol. 2012. 11: 217-24
5. Amin-Hanjani S, Ogilvy CS, Candia GJ, Lyons S, Chapman PH. Stereotactic radiosurgery for cavernous malformations: Kjellberg's experience with proton beam therapy in 98 cases at the Harvard Cyclotron. Neurosurgery. 1998. 42: 1229-36
6. Barker FG, Amin-Hanjani S, Butler WE, Lyons S, Ojemann RG, Chapman PH. Temporal clustering of hemorrhages from untreated cavernous malformations of the central nervous system. Neurosurgery. 2001. 49: 15-24
7. Baumann CR, Schuknecht B, Lo Russo G, Cossu M, Citterio A, Andermann F. Seizure outcome after resection of cavernous malformations is better when surrounding hemosiderin-stained brain also is removed. Epilepsia. 2006. 47: 563-6
8. Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005. 76: 42-51
9. Bertalanffy H, Benes L, Miyazawa T, Alberti O, Siegel AM, Sure U. Cerebral cavernomas in the adult. Review of the literature and analysis of 72 surgically treated patients. Neurosurgical Rev. 2002. 25: 1-53
10. Bicknell JM, Carlow TJ, Kornfeld M, Stovring J, Turner P. Familial cavernous angiomas. Arch Neurol. 1978. 35: 746-9
11. Borikova AL, Dibble CF, Sciaky N, Welch CM, Abell AN, Bencharit S. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010. 285: 11760-4
12. Cavalcanti DD, Kalani MY, Martirosyan NL, Eales J, Spetzler RF, Preul MC. Cerebral cavernous malformations: From genes to proteins to disease. J Neurosurg. 2012. 116: 122-32
13. Chang EF, Gabriel RA, Potts MB, Berger MS, Lawton MT. Supratentorial cavernous malformations in eloquent and deep locations: Surgical approaches and outcomes. Clinical article. J Neurosurg. 2011. 114: 814-27
14. Chang EF, Gabriel RA, Potts MB, Garcia PA, Barbaro NM, Lawton MT. Seizure characteristics and control after microsurgical resection of supratentorial cerebral cavernous malformations. Neurosurgery. 2009. 65: 31-7
15. Chen L, Zhao Y, Zhou L, Zhu W, Pan Z, Mao Y. Surgical strategies in treating brainstem cavernous malformations. Neurosurgery. 2011. 68: 609-20
16. Clark JV. Familial occurrence of cavernous angiomata of the brain. J Neurol Neurosurg Psychiatry. 1970. 33: 871-6
17. Clatterbuck RE, Moriarity JL, Elmaci I, Lee RR, Breiter SN, Rigamonti D. Dynamic nature of cavernous malformations: A prospective magnetic resonance imaging study with volumetric analysis. J Neurosurg. 2000. 93: 981-6
18. Cohen-Inbar O, Xu Z, Sheehan JP. Focused ultrasound-aided immunomodulation in glioblastoma multiforme: A therapeutic concept. J Ther Ultrasound. 2016. 4: 2-
19. Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998. 7: 1851-8
20. Dashti SR, Hoffer A, Hu YC, Selman WR. Molecular genetics of familial cerebral cavernous malformations. Neurosurg Focus. 2006. 21: e2-
21. Davenport WJ, Siegel AM, Dichgans J, Drigo P, Mammi I, Pereda P. CCM1 gene mutations in families segregating cerebral cavernous malformations. Neurology. 2001. 56: 540-3
22. Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004. 74: 326-37
23. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006. 60: 550-6
24. Duckworth EA. Modern management of brainstem cavernous malformations. Neurol Clin. 2010. 28: 887-98
25. Dupre N, Verlaan DJ, Hand CK, Laurent SB, Turecki G, Davenport WJ. Linkage to the CCM2 locus and genetic heterogeneity in familial cerebral cavernous malformation. Can J Neurol Sci. 2003. 30: 122-8
26. Eerola I, McIntyre B, Vikkula M. Identification of eight novel 5’- exons in cerebral capillary malformation gene-1 (CCM1) encoding KRIT1. Biochim Biophys Acta. 2001. 1517: 464-7
27. Eisa-Beygi S, Wen XY, Macdonald RL. A call for rigorous study of statins in resolution of cerebral cavernous malformation pathology. Stroke. 2014. 45: 1859-61
28. Englot DJ, Han SJ, Lawton MT, Chang EF. Predictors of seizure freedom in the surgical treatment of supratentorial cavernous malformations. J Neurosurg. 2011. 115: 1169-74
29. Flemming KD, Link MJ, Christianson TJ, Brown RD, Jr . Prospective hemorrhage risk of intracerebral cavernous malformations. Neurology. 2012. 78: 632-6
30. Gault J, Awad IA, Recksiek P, Shenkar R, Breeze R, Handler M. Cerebral cavernous malformations: Somatic mutations in vascular endothelial cells. Neurosurgery. 2009. 65: 138-44
31. Gault J, Shenkar R, Recksiek P, Awad IA. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke. 2005. 36: 872-4
32. Ghanouni P, Pauly KB, Elias WJ, Henderson J, Sheehan J, Monteith S. Transcranial MRI-Guided Focused Ultrasound: A Review of the Technologic and Neurologic Applications. AJR Am J Roentgenol. 2015. 205: 150-9
33. Glading AJ, Ginsberg MH. Rap1 and its effector KRIT1/CCM1 regulate beta-catenin signaling. Dis Model Mech. 2010. 3: 73-83
34. Gross BA, Du R, Orbach DB, Scott RM, Smith ER. The natural history of cerebral cavernous malformations in children. J Neurosurg Pediatr. 2015. p. 1-6
35. Gross BA, Lin N, Du R, Day AL. The natural history of intracranial cavernous malformations. Neurosurgical focus. 2011. 30: E24-
36. Hammen T, Romstock J, Dorfler A, Kerling F, Buchfelder M, Stefan H. Prediction of postoperative outcome with special respect to removal of hemosiderin fringe: A study in patients with cavernous haemangiomas associated with symptomatic epilepsy. Seizure. 2007. 16: 248-53
37. Hayman LA, Evans RA, Ferrell RE, Fahr LM, Ostrow P, Riccardi VM. Familial cavernous angiomas: Natural history and genetic study over a 5-year period. Am J Med Genet. 1982. 11: 147-60
38. Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995. 377: 695-701
39. Karlsson B, Kihlstrom L, Lindquist C, Ericson K, Steiner L. Radiosurgery for cavernous malformations. J Neurosurg. 1998. 88: 293-7
40. Kidd HA, Cumings JN. Cerebral angiomata in an Icelandic family. Lancet. 1947. 1: 747-
41. Kivelev J, Niemela M, Hernesniemi J. Treatment strategies in cavernomas of the brain and spine. J Clin Neurosci. 2012. 19: 491-7
42. Knudson AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971. 68: 820-3
43. Kondziolka D, Lunsford LD, Kestle JR. The natural history of cerebral cavernous malformations. J Neurosurg. 1995. 83: 820-4
44. Kotagal P, Luüders H.editorsThe Epilepsies: Etiologies and Prevention. San Diego: Academic Press; 1999. p.
45. Kraemer DL, Awad IA. Vascular malformations and epilepsy: Clinical considerations and basic mechanisms. Epilepsia. 1994. 35: S30-43
46. Kraemer DL, Griebel ML, Lee N, Friedman AH, Radtke RA. Surgical outcome in patients with epilepsy with occult vascular malformations treated with lesionectomy. Epilepsia. 1998. 39: 600-7
47. Labauge P, Laberge S, Brunereau L, Levy C, Tournier-Lasserve E. Hereditary cerebral cavernous angiomas: Clinical and genetic features in 57 French families. Societe Francaise de Neurochirurgie. Lancet. 1998. 352: 1892-7
48. Li D, Hao SY, Tang J, Xiao XR, Jia GJ, Wu Z. Clinical course of untreated pediatric brainstem cavernous malformations: Hemorrhage risk and functional recovery. J Neurosurg Pediatr. 2014. 13: 471-83
49. Li DY, Whitehead KJ. Evaluating strategies for the treatment of cerebral cavernous malformations. Stroke. 2010. 41: S92-4
50. Ma X, Zhao H, Shan J, Long F, Chen Y, Chen Y. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007. 18: 1965-78
51. Marchuk DA, Gallione CJ, Morrison LA, Clericuzio CL, Hart BL, Kosofsky BE. A locus for cerebral cavernous malformations maps to chromosome 7q in two families. Genomics. 1995. 28: 311-4
52. Matos P, Skaug J, Marques B, Beck S, Verissimo F, Gespach C. Small GTPase Rac1: Structure, localization, and expression of the human gene. Biochem Biophys Res Commun. 2000. 277: 741-51
53. McCracken DJ, Willie JT, Fernald BA, Saindane AM, Drane DL, Barrow DL. Magnetic Resonance Thermometry-Guided Stereotactic Laser Ablation of Cavernous Malformations in Drug-Resistant Epilepsy: Imaging and Clinical Results. Neurosurgery. 2015. p.
54. Moriarity JL, Wetzel M, Clatterbuck RE, Javedan S, Sheppard JM, Hoenig-Rigamonti K. The natural history of cavernous malformations: A prospective study of 68 patients. Neurosurgery. 1999. 44: 1166-71
55. Nimjee SM, Powers CJ, Bulsara KR. Review of the literature on de novo formation of cavernous malformations of the central nervous system after radiation therapy. Neurosurg Focus. 2006. 21: e4-
56. Patel P, Patel NV, Danish SF. Intracranial MR-guided laser-induced thermal therapy: Single-center experience with the Visualase thermal therapy system. J Neurosurg. 2016. p. 1-8
57. Pham M, Gross BA, Bendok BR, Awad IA, Batjer HH. Radiosurgery for angiographically occult vascular malformations. Neurosurg Focus. 2009. 26: E16-
58. Pizon V, Chardin P, Lerosey I, Olofsson B, Tavitian A. Human cDNAs rap1 and rap2 homologous to the Drosophila gene Dras3 encode proteins closely related to ras in the ‘effector’ region. Oncogene. 1988. 3: 201-4
59. Porter PJ, Willinsky RA, Harper W, Wallace MC. Cerebral cavernous malformations: Natural history and prognosis after clinical deterioration with or without hemorrhage. Journal of neurosurgery. 1997. 87: 190-7
60. Porter RW, Detwiler PW, Han PP, Spetzler RF. Stereotactic radiosurgery for cavernous malformations: Kjellberg's experience with proton beam therapy in 98 cases at the Harvard Cyclotron. Neurosurgery. 1999. 44: 424-5
61. Pozzati E, Acciarri N, Tognetti F, Marliani F, Giangaspero F. Growth, subsequent bleeding, and de novo appearance of cerebral cavernous angiomas. Neurosurgery. 1996. 38: 662-9
62. Ramina R, Mattei TA, de Aguiar PH, Meneses MS, Ferraz VR, Aires R. Surgical management of brainstem cavernous malformations. Neurol Sci. 2011. 32: 1013-28
63. Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT. Cerebral cavernous malformations. Incidence and familial occurrence. N Eng J Med. 1988. 319: 343-7
64. Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991. 75: 709-14
65. Ruggieri R, Bender A, Matsui Y, Powers S, Takai Y, Pringle JR. RSR1, a ras-like gene homologous to Krev-1 (smg21A/rap1A): Role in the development of cell polarity and interactions with the Ras pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1992. 12: 758-66
66. Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999. 8: 2325-33
67. Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997. 15: 1043-9
68. Sheehan J. Transcranial ultrasound for arteriovenous malformations: Something old is new again. World Neurosurg. 2012. 77: 269-70
69. Stavrou I, Baumgartner C, Frischer JM, Trattnig S, Knosp E. Long-term seizure control after resection of supratentorial cavernomas: A retrospective single-center study in 53 patients. Neurosurgery. 2008. 63: 888-96
70. Stefan H, Hammen T. Cavernous haemangiomas, epilepsy and treatment strategies. Acta Neurol Scand. 2004. 110: 393-7
71. Stockton RA, Shenkar R, Awad IA, Ginsberg MH. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010. 207: 881-96
72. Tomlinson FH, Houser OW, Scheithauer BW, Sundt TM, Okazaki H, Parisi JE. Angiographically occult vascular malformations: A correlative study of features on magnetic resonance imaging and histological examination. Neurosurgery. 1994. 34: 792-9
73. Van Gompel JJ, Rubio J, Cascino GD, Worrell GA, Meyer FB. Electrocorticography-guided resection of temporal cavernoma: Is electrocorticography warranted and does it alter the surgical approach?. J Neurosurg. 2009. 110: 1179-85
74. Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998. 93: 741-53
75. Wang Z, Qian C, Shi L, Wang L, Zhang J, Wang Y. Surgery Approaches to Brainstem Cavernous Malformations. J Craniofac Surg. 2015. 26: e577-80
76. Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development. 2004. 131: 1437-48
77. Wienecke R, Maize JC, Reed JA, de Gunzburg J, Yeung RS, DeClue JE. Expression of the TSC2 product tuberin and its target Rap1 in normal human tissues. Am J Pathol. 1997. 150: 43-50
78. Wilfong AA, Curry DJ. Hypothalamic hamartomas: Optimal approach to clinical evaluation and diagnosis. Epilepsia. 2013. 54: 109-14
79. Yadla S, Jabbour PM, Shenkar R, Shi C, Campbell PG, Awad IA. Cerebral cavernous malformations as a disease of vascular permeability: From bench to bedside with caution. Neurosurg Focus. 2010. 29: E4-
80. Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP. The natural history of familial cavernous malformations: Results of an ongoing study. J Neurosurg. 1994. 80: 422-32
81. Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. KRIT1 association with the integrin-binding protein ICAP-1: A new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet. 2002. 11: 389-96
82. Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL. CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005. 14: 2521-31
83. Zhang J, Clatterbuck RE, Rigamonti D, Chang DD, Dietz HC. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum Mol Genet. 2001. 10: 2953-60
84. Zhang J, Clatterbuck RE, Rigamonti D, Dietz HC. Cloning of the murine Krit1 cDNA reveals novel mammalian 5’ coding exons. Genomics. 2000. 70: 392-5
85. Zhang N, Pan L, Wang BJ, Wang EM, Dai JZ, Cai PW. Gamma knife radiosurgery for cavernous hemangiomas. J Neurosurg. 2000. 93: S74-7

