

- Departments of Neurosurgery, Karolinska University Hospital, Stockholm, Sweden,
- Department of Oncology, Royal Berkshire NHS Foundation Trust, Reading, United Kingdom,
- Department of Neurosurgery, Bezmialem Vakif University Medical School, İstanbul, Turkey.
- Departments of Medical Radiation Physics and Nuclear Medicine, Karolinska University Hospital, Stockholm, Sweden,
- Departments of Neuropathology, Karolinska University Hospital, Stockholm, Sweden,
Correspondence Address:
Georges Sinclair
Departments of Neuropathology, Karolinska University Hospital, Stockholm, Sweden,
DOI:10.25259/SNI_112_2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Georges Sinclair, Martin Olsson, Hamza Benmakhlouf, Yahya Al-saffar, Philippa Johnstone, Mustafa Aziz Hatiboglu, Alia Shamikh. Pituitary carcinomas: Rare and challenging. 09-Aug-2019;10:161
How to cite this URL: Georges Sinclair, Martin Olsson, Hamza Benmakhlouf, Yahya Al-saffar, Philippa Johnstone, Mustafa Aziz Hatiboglu, Alia Shamikh. Pituitary carcinomas: Rare and challenging. 09-Aug-2019;10:161. Available from: http://surgicalneurologyint.com/surgicalint-articles/9569/
Date of Submission
11-Mar-2019
Date of Acceptance
28-May-2019
Date of Web Publication
09-Aug-2019
Abstract
Background: Pituitary carcinomas (PCs) are defined as adenohypophyseal tumors with metastatic activity within and outside the boundaries of the central nervous system (CNS). The condition is rare and therefore seldom reported; most lesions are hormone producing and have a tendency for complex evolution. As such, the management of PCs remains difficult. We present an illustrative case of PC with a brief review of the recent medical literature.
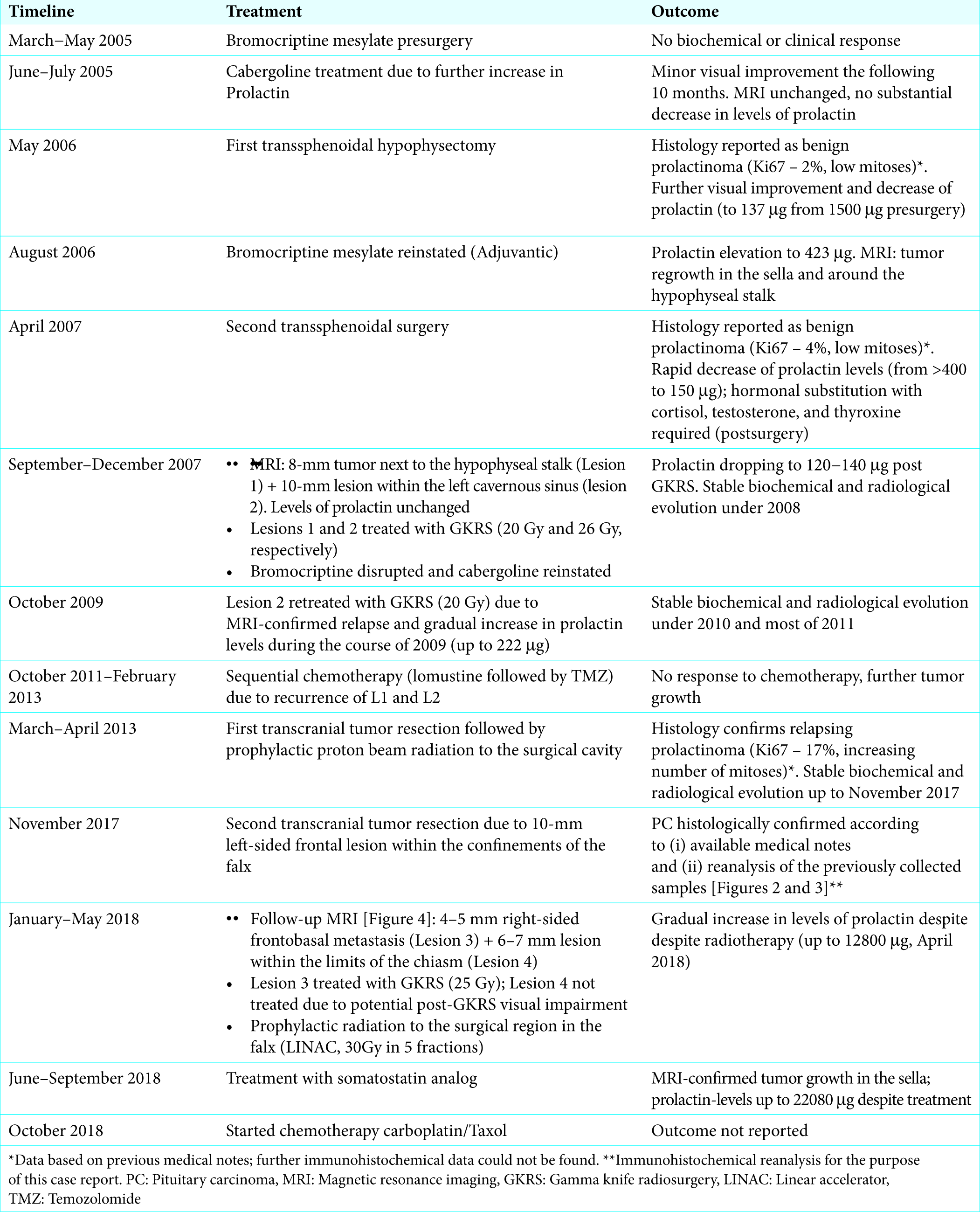
Case Description: A 58-year-old patient was diagnosed with prolactinoma in 2005. The ensuing biochemical and radiological evolution proved contentious; local tumor control was never fully achieved despite multimodal management including pharmacological treatment, repeated resections, and radiotherapy. In late 2017, the patient developed metastatic lesions within the confinements of the CNS requiring further surgical interventions, high-dose radiation, and systemic treatment.
Conclusion: As it was the case in our patient, PCs require tailored, multimodal treatments according to the degree of infiltration, site of invasion, and hormone status. Further studies are necessary to understand the mechanisms promoting “extra-sellar” activity, particularly at distant sites; the identification of biomarkers exposing the risk of PC remains a crucial aspect of diagnostics, prevention and future customized therapies.
Keywords: Adenoma, Central nervous system, Metastatic activity, Pituitary carcinoma
INTRODUCTION
Pituitary carcinomas (PCs) are rare malignant neoplasms, accounting for approximately 0.12% of adenohypophyseal tumors and 6% of local invasive adenomas.[
CASE DESCRIPTION
We present a case of a previously healthy, 58-year-old male patient, who developed bitemporal hemianopsia in the first few months of 2005. The ensuing radiological assessment revealed a 22 mm × 20 mm × 15 mm pituitary mass with chiasmatic/bilateral optic nerve upward dislocation [
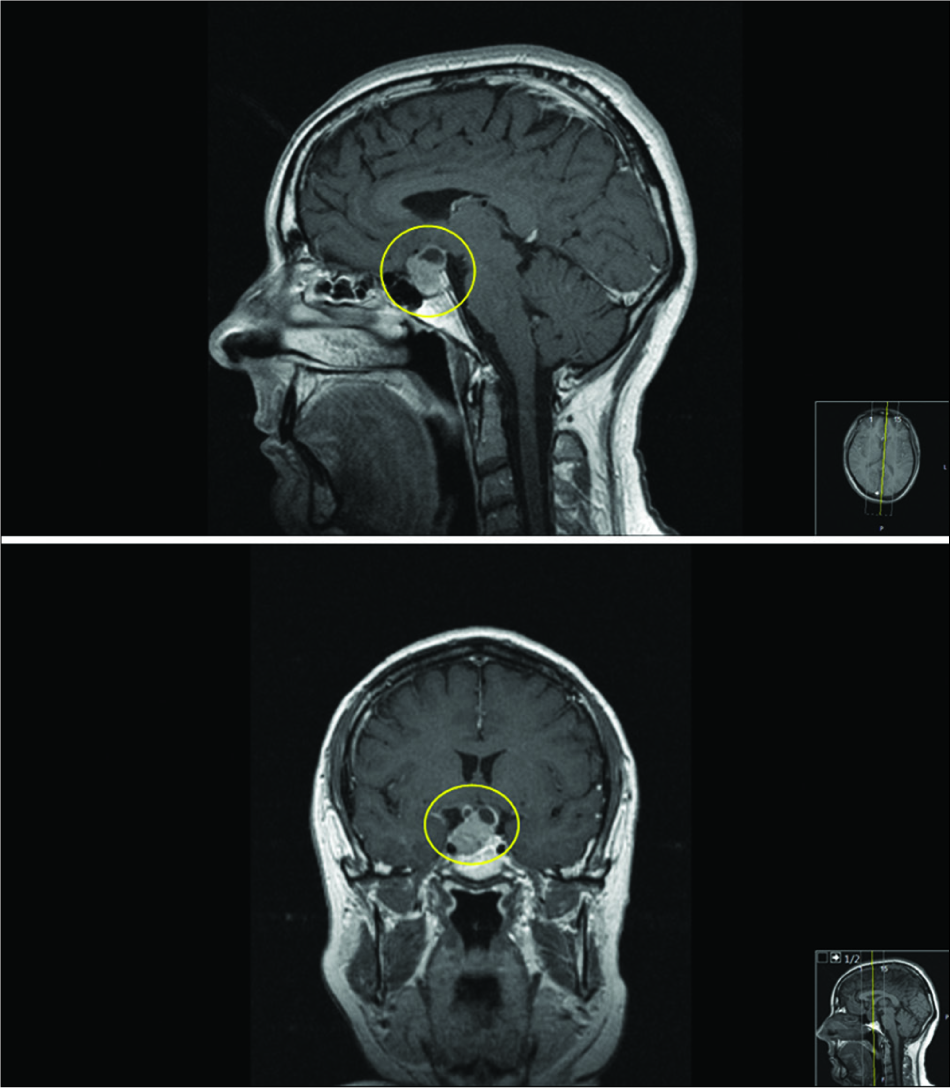
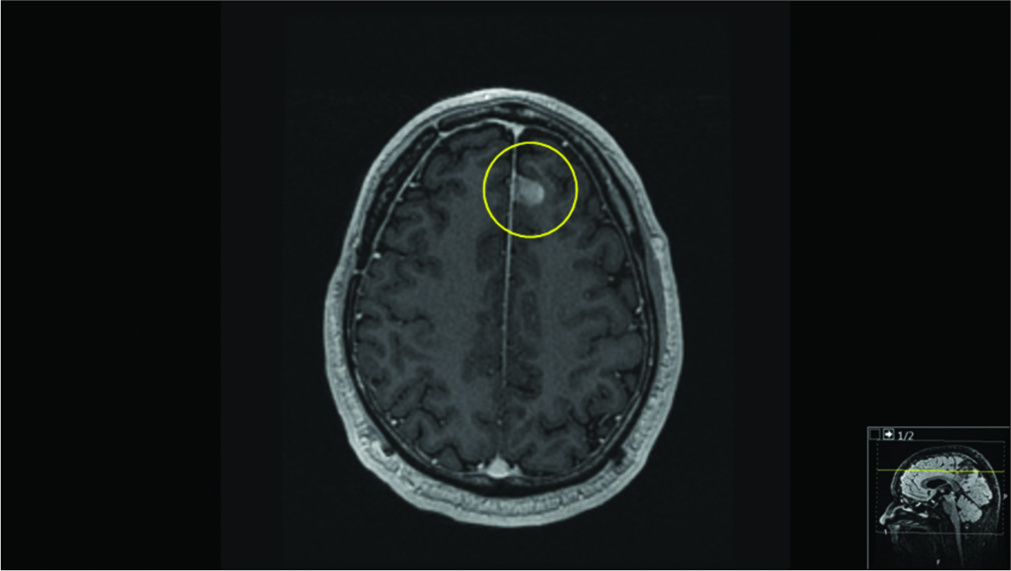
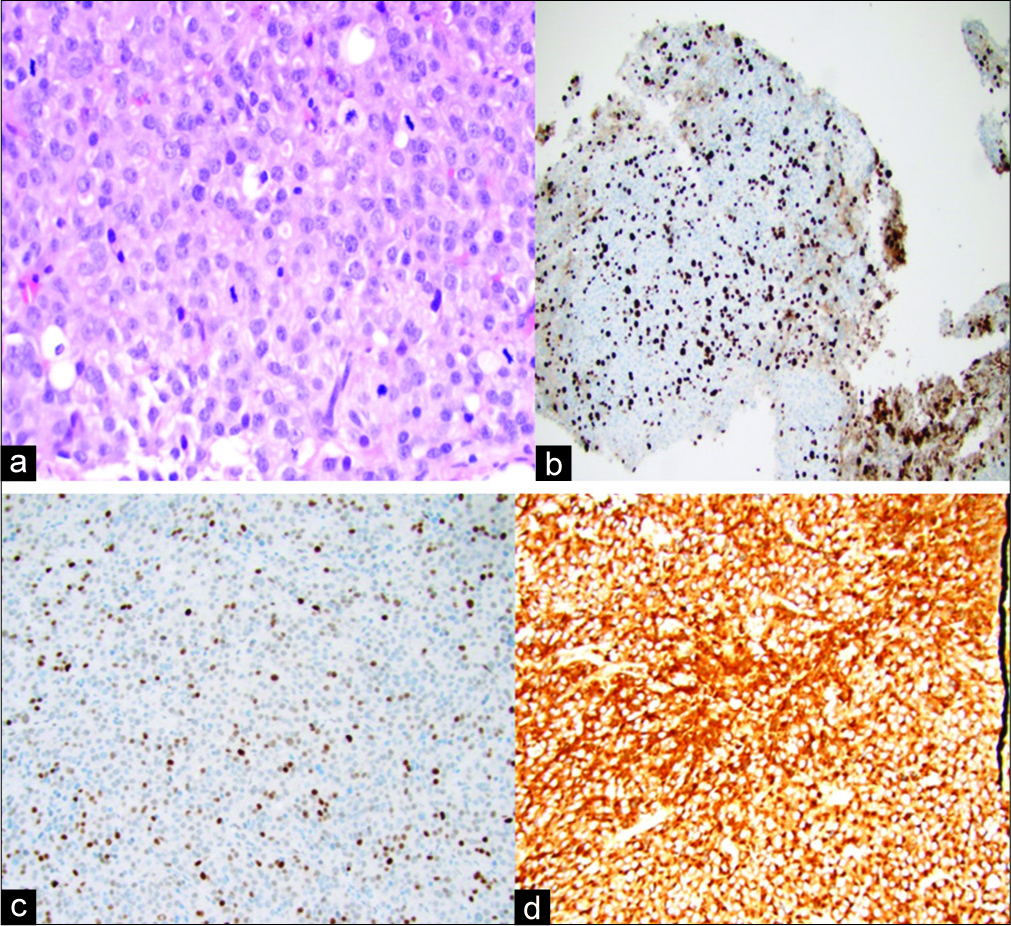
Figure 3:
(×400). Microscopic reassessment of samples from second metastasectomy (November 2017) for the purpose of this article (a) H&E staining: high mitotic activity in a population of large cells with atypical nuclei and prominent nucleoli. (b) KI67: high proliferation (35%). (c) Overexpression of P53 limited to a few tumor cells (anti-p53 antibody). (d) Diffuse Immunostaining for prolactin. Of note, thyroid stimulating hormone, growth hormone, adrenocorticotropic hormone, CK-AE1AE3 proved negative. Samples from the first metastasectomy were not made available for reanalysis.
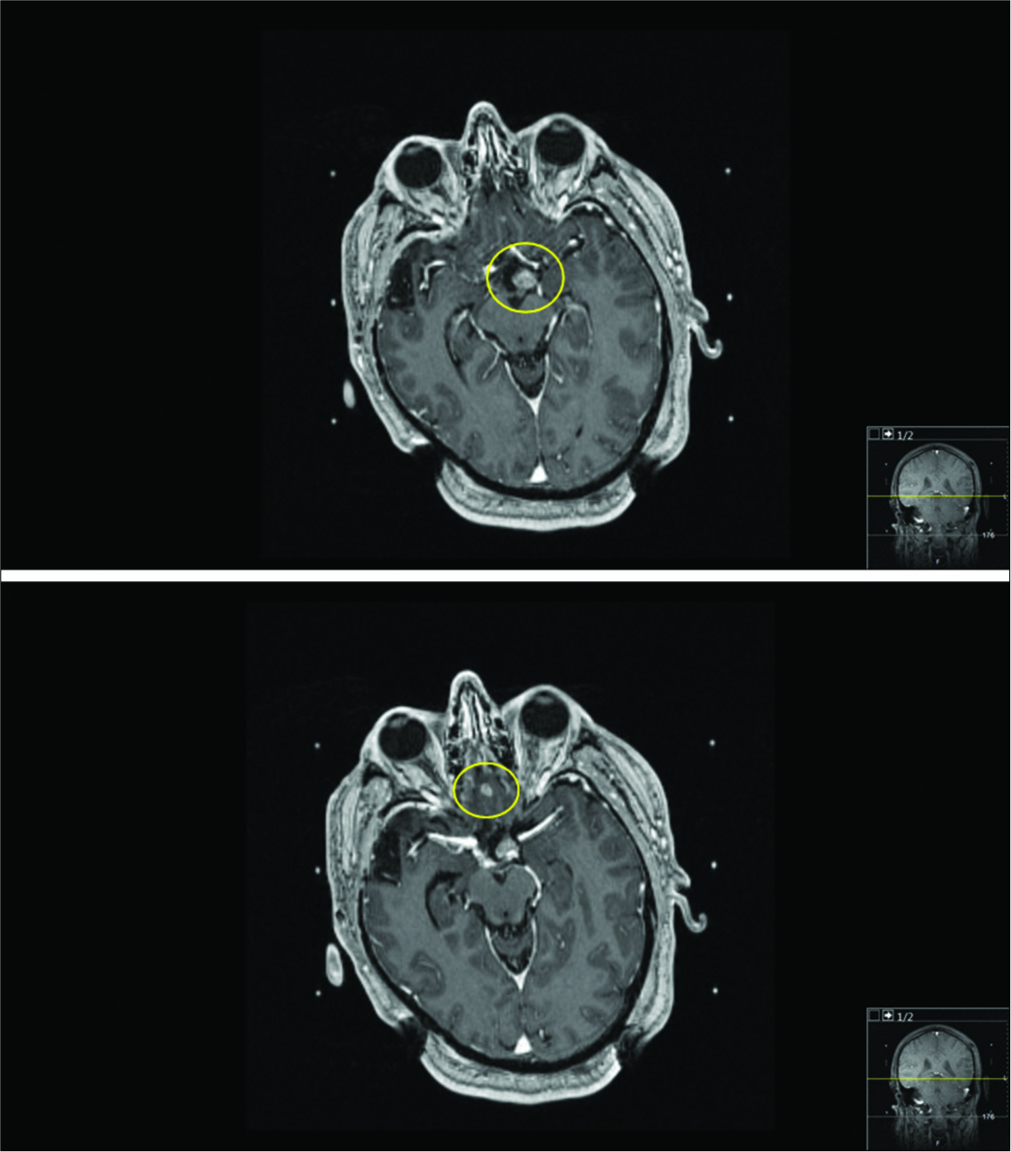
DISCUSSION
General aspects
Hypophyseal tumors account for 15% of all intracranial tumors;[
Clinical characteristics
PC activity prevails in central nervous system (CNS) locations although other sites of dissemination such as the liver, bone, heart, ovaries, and lymph nodes have also been reported.[
Immunohistochemistry
As is the case here, a thorough microscopic tumor evaluation from the primary and metastatic sites remains crucial in confirming diagnosis and assessing best treatment options. Histologically, PC lesions may look like typical adenomas but may also display marked pleomorphism and frequent mitoses.[
Treatment modalities of manifest PC
The treatment of PCs remains multimodal and includes surgical resection (transsphenoidal and transcranial), linear accelerator (LINAC)- and proton-beam- based fractionated radiotherapy, single-dose GKRS, chemotherapy, immunotherapy, and the use of other pharmacological agents targeting hormone production itself.[
CONCLUSION
PCs are rare neoplasms with contentious metastatic evolution and require multimodal, tailored treatments. Unfortunately, despite modern medical technology, the prognosis remains poor. Although the incidence of PCs is low, further studies are necessary to understand the proliferative mechanisms leading to local invasion and metastatic activity. The identification of prognostic biomarkers for risk stratification and treatment response remains necessary in terms of prevention of PCs and future selected therapies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
References
1. AbdelBaki MS, Waguespack SG, Salceda V, Jones J, Stapleton SL, Baskin DS. Significant response of pituitary carcinoma to carboplatin, leucovorin and fluorouracil chemotherapy: A pediatric case report and review of the literature. J Neurooncol. 2017. 135: 213-5
2. Balili I, Sullivan S, Mckeever P, Barkan A. Pituitary carcinoma with endolymphatic sac metastasis. Pituitary. 2014. 17: 210-3
3. Cerovac V, Monteserin-Garcia J, Rubinfeld H, Buchfelder M, Losa M, Florio T. The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res. 2010. 70: 666-74
4. Dudziak K, Honegger J, Bornemann A, Horger M, Mussig K. Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course case report and review of the literature. J Clin Endocrinol Metab. 2011. 96: 2665-9
5. Galland F, Lacroix L, Saulnier P, Dessen P, Meduri G, Bernier M. Differential gene expression profiles of invasive and non-invasive non-functioning pituitary adenomas based on microarray analysis. Endocr Relat Cancer. 2010. 17: 361-71
6. Jouanneau E, Wierinckx A, Ducray F, Favrel V, Borson-Chazot F, Honnorat J. New targeted therapies in pituitary carcinoma resistant to temozolomide. Pituitary. 2012. 15: 37-43
7. Landman RE, Horwith M, Peterson RE, Khandji AG, Wardlaw SL. Long-term survival with ACTH-secreting carcinoma of the pituitary: A case report and review of the literature. J Clin Endocrinol Metab. 2002. 87: 3084-9
8. Lloyd RV, Osamura YR, Klöppel G, Rosai J, Organization WH.editorsWHO Classification of Tumours of Endocrine Organs. Lyon: IARC. Press; 2017. p.
9. Maira G, Doglietto F. Pituitary carcinoma: A devastating disease in need of an earlier diagnosis and of effective therapies. World Neurosurg. 2013. 80: e143-5
10. Morokuma H, Ando T, Hayashida T, Horie I, Inoshita N, Murata F. A case of nonfunctioning pituitary carcinoma that responded to temozolomide treatment. Case Rep Endocrinol. 2012. 2012: 645914-
11. Park KS, Hwang JH, Hwang SK, Kim S, Park SH. Pituitary carcinoma with fourth ventricle metastasis: Treatment by excision and gamma-knife radiosurgery. Pituitary. 2014. 17: 514-8
12. Philippon M, Morange I, Barrie M, Barlier A, Taieb D, Dufour H. Long-term control of a MEN1 prolactin secreting pituitary carcinoma after temozolomide treatment. Ann Endocrinol (Paris). 2012. 73: 225-9
13. Phillips J, East HE, French SE, Melcescu E, Hamilton RD, Nicholas WC. What causes a prolactinoma to be aggressive or to become a pituitary carcinoma. ? Hormones (Athens). 2012. 11: 477-82
14. Raverot G, Sturm N, de Fraipont F, Muller M, Salenave S, Caron P. Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: A French multicenter experience. J Clin Endocrinol Metab. 2010. 95: 4592-9
15. Sansur CA, Oldfield EH. Pituitary carcinoma. Semin Oncol. 2010. 37: 591-3
16. Scheithauer BW, Kovacs K, Nose V, Lombardero M, Osamura YR, Lloyd RV. Multiple endocrine neoplasia Type 1 associated thyrotropin-producing pituitary carcinoma: Report of a probable de novo example. Hum Pathol. 2009. 40: 270-8
17. Shastri BR, Nanda A, Fowler M, Levine SN. Adrenocorticotropic hormone-producing pituitary carcinoma with intracranial metastases. World Neurosurg. 2013. 79: 404-6
18. Touma W, Hoostal S, Peterson RA, Wiernik A, SantaCruz KS, Lou E. Successful treatment of pituitary carcinoma with concurrent radiation, temozolomide, and bevacizumab after resection. J Clin Neurosci. 2017. 41: 75-7
19. Tufton N, Roncaroli F, Hadjidemetriou I, Dang MN, Dénes J, Guasti L. Pituitary carcinoma in a patient with an SDHB mutation. Endocr Pathol. 2017. 28: 320-5
20. Zemmoura I, Wierinckx A, Vasiljevic A, Jan M, Trouillas J, François P. Aggressive and malignant prolactin pituitary tumors: Pathological diagnosis and patient management. Pituitary. 2013. 16: 515-22

