

- Department of Neurosurgery, Kenneth R. Peak Brain and Pituitary Center, Houston Methodist Neurological Institute, Houston, TX, USA
Correspondence Address:
David S. Baskin
Department of Neurosurgery, Kenneth R. Peak Brain and Pituitary Center, Houston Methodist Neurological Institute, Houston, TX, USA
DOI:10.4103/2152-7806.166176
Copyright: © 2015 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Huang M, Steele WJ, Baskin DS. Primary central nervous system vasculitis preceded by granulomatous hypophysitis: Case report with a review of the literature. Surg Neurol Int 28-Sep-2015;6:
How to cite this URL: Huang M, Steele WJ, Baskin DS. Primary central nervous system vasculitis preceded by granulomatous hypophysitis: Case report with a review of the literature. Surg Neurol Int 28-Sep-2015;6:. Available from: http://surgicalneurologyint.com/surgicalint_articles/primary-central-nervous-system-vasculitis-preceded-by/
Abstract
Background:Primary central nervous system (CNS) vasculitis is an idiopathic inflammatory process that selectively affects CNS vasculature without a systemic inflammatory response, and causes luminal obstruction with resultant ischemia of recipient tissue. Its varying clinical symptoms and signs depend on the caliber of vessels involved and distribution and location of the affected structures. Granulomatous hypophysitis (GH) is an autoimmune inflammatory process typically affecting women, and usually presents with hypopituitarism, and at times, diabetes insipidus, and/or visual loss. Both entities are rare CNS diseases, which, to our knowledge, have never been previously reported in the same patient.
Case Description:We present a unique case of chronic progressive primary CNS vasculitis causing limbic encephalopathy in a 30-year-old male with only a history of medication-controlled hypertension. He initially presented 4 months prior with nonspecific neurological complaints and was found to have a homogenously enhancing and enlarged pituitary, which was biopsy proven to be GH.
Conclusion:This rather unique presentation highlights the need to maintain a high index of suspicion for underlying PCNS vasculitis in a patient who does not fit the typical demographic for isolated GH.
Keywords: Central nervous system, hypophysitis, vasculitis, primary
INTRODUCTION
Vasculitis is characterized by blood vessel inflammation that precipitates luminal occlusion and subsequent infarction. Clinical manifestations of vasculitis, which are often variable and nonspecific, depend on the caliber of affected vessel (s), the extent of local involvement, and the type of target organ (s). Vasculitis of the central nervous system (CNS) is most commonly secondary to a systemic process with collateral CNS involvement. Primary CNS vasculitis (PCNSV) is a poorly understood, rare, and complex disease with protean clinical manifestations that occurs in the absence of a systemic inflammatory process.[
Granulomatous hypophysitis (GH) is a rare autoimmune inflammatory disorder of the pituitary gland typically affecting middle-aged women, and may be a later manifestation of lymphocytic hypophysitis (LYH), which usually affects younger peripartum females. The cases of GH and LYH involving nonpregnant women and men have been reported. Clinically, both conditions can involve the anterior and posterior pituitary, which can clinically manifest with their respective endocrine dysfunction syndromes.[
We present a unique case of progressive PCNSV causing limbic encephalopathy in a 29-year-old male who initially presented with isolated GH. The case is also of interest because it mimicked herpes encephalitis both clinically and radiologically.
CASE REPORT
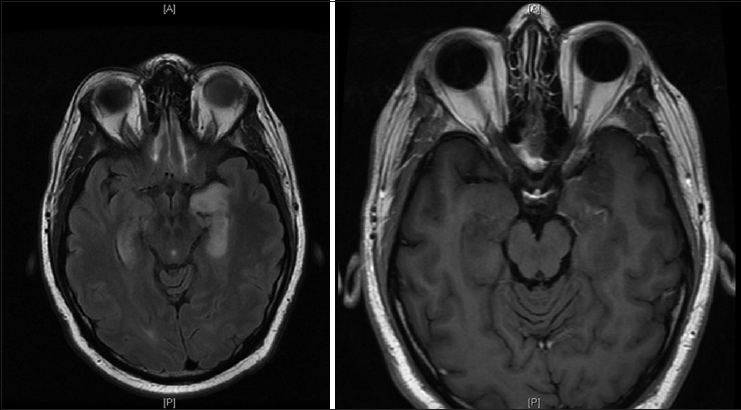
A 29-year-old male initially presented to an otolaryngologist's office for evaluation of the acute onset of left-sided hearing loss. His past medical history was significant only for obesity with a body mass index 34, hypertension, and presumptive shingles on his left flank treated with valacyclovir, although no exanthema had ever appeared. He endorsed occasional alcohol but no smoking or illicit drug usage. The patient reported a 2 weeks history of headache, nausea, and vomiting was accompanied by photophobia. He had been to an emergency room, and anti-nausea and pain medications had been prescribed. While at the office, he received a local steroid injection, which provided no resolution of his symptoms. A subsequent magnetic resonance imaging (MRI) obtained at that time showed a diffusely enhancing and enlarged pituitary gland [
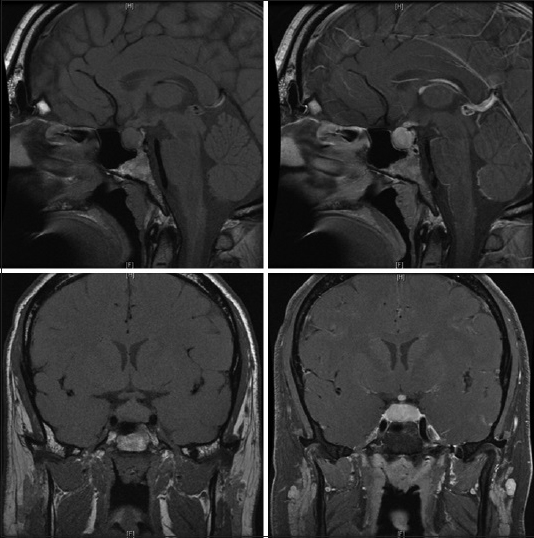
Figure 1
Pre- and post-contrast magnetic resonance images show the pituitary to be diffusely enlarged with homogenous enhancement, measuring 1.3 cm × 2.0 cm × 1.1 cm in cranial-caudal, transverse, and anterior-posterior dimensions, respectively. The pituitary stalk is markedly thickened and intensely enhancing
The patient was seen in the clinic by senior author and was subsequently admitted for transsphenoidal exploration and biopsy of the pituitary mass. Hormonal testing at that time was significant for thyroid-stimulating hormone level of 0.01 uIU/ml, luteinizing hormone of 0.8 mIU/ml, testosterone < 3, insulin-like growth factor-1 of 319, dehydroepiandrosterone at 13.2 ug/dl. The patient tolerated the procedure well, and intraoperative frozen pathology showed rubbery anterior pituitary gland with diffuse lymphocytic infiltration consistent with lymphocytic adenophypophysitis.
The final pathology, however, was interpreted to represent GH. Special stains for microorganisms were negative. Microscopically, cytologic preparations adenosine triphosphate showed predominantly blood with scattered lymphocytes and macrophages. Frozen (AFS) and permanent sections showed a pituitary gland with marked noncaseating granulomatous inflammation and diffused lymphocytic infiltrate. Immunostains such as CD68 confirmed the presence of histiocytes and CD3 and CD20 immunostains showed the lymphocytes to be a reactive population. Cytokeratin immunostains highlighted the pituitary gland and the intact acinar architecture in the background of inflammation. Special stains for fungal Gomori methenamine silver (GMS), bacterial (gram), and mycobacterial (Fite) organisms were negative. Cerebrospinal fluid (CSF) analysis was negative for malignancy, and immunohistochemical staining for herpes simplex virus (HSV1–2) was negative. The patient was discharged on postoperative day 1 neurologically intact and prescribed thyroid, testosterone, and pulsed high-dose oral steroid regimen. The patient's condition improved, and a subsequent MRI scan showed a decrease in size of pituitary back to a more normal configuration [
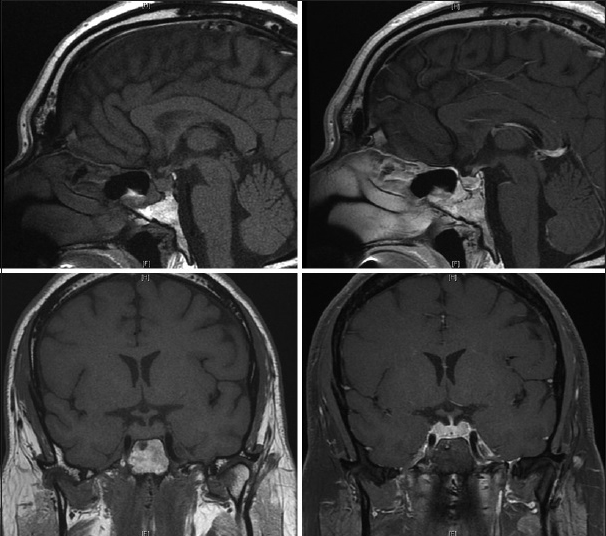
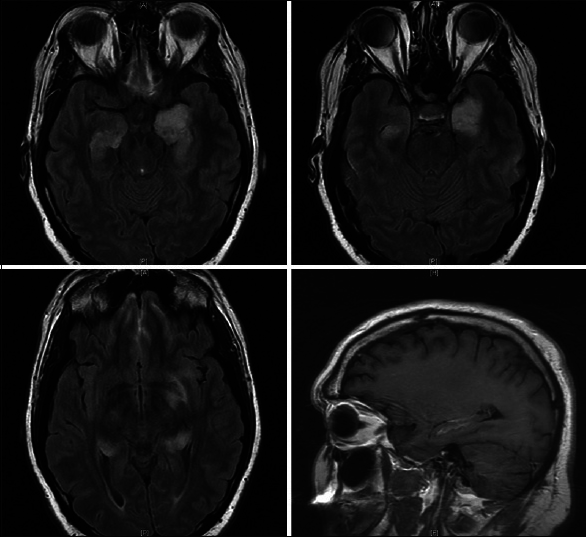
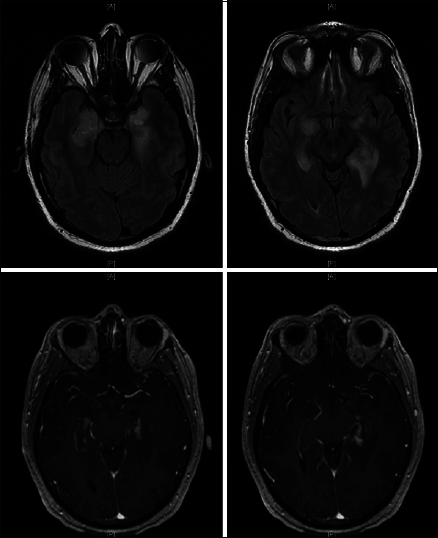
Approximately, 3 months after discharge, the patient developed fatigue, malaise, and progressive short-term memory deficits that progressed rapidly over a course of 2 weeks. He retained his remote memory and was able to recall name, birthday, and president without difficulty. There was no fever, weight loss, chills, vision changes, slurred speech, facial droop, weakness, numbness, issues with gait, or other focal deficits except for his prior hearing loss in his left ear. He was tapering his steroid medications. The patient was readmitted to the hospital for concern of HSV encephalitis. MRI showed T2 signal changes, as well as bilateral contrast enhancement, in the medial temporal lobes [
A lumbar puncture was performed, which was negative for an infectious etiology including routine, acid-fast bacilli (AFB), and fungal cultures, cryptococcus antigen, HSV 1,2 HIV 1,2 Epstein–Barr virus (EBV), West Nile, venereal disease research laboratory, varizella zoster, and Borrelia burgdorferi. Flow cytometry showed no clonal B cells [
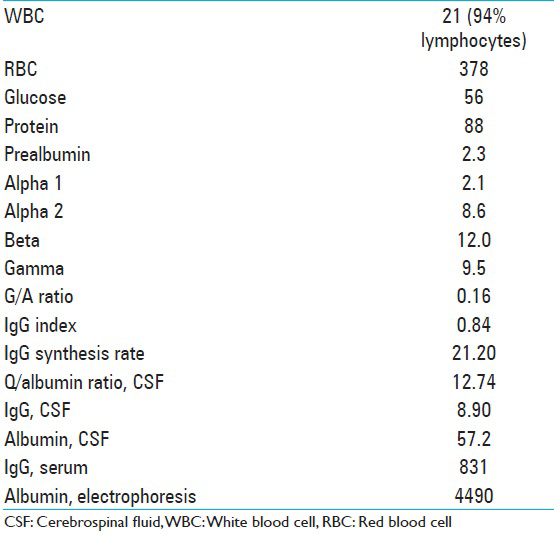
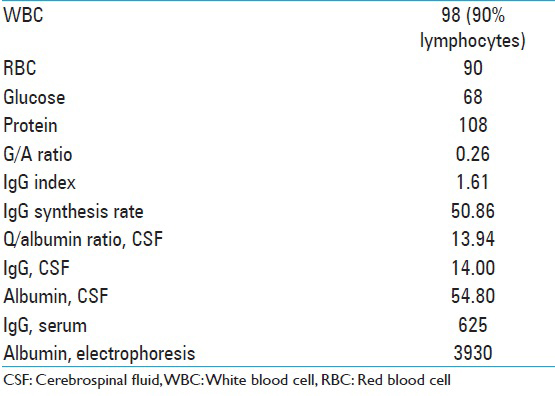
Fifty percent of the cell populations were T cells by CD5 immunotyping, 8% mature B cells, with no light chain restriction or significant co-expression of CD5 or CD10, and <1% plasma cells. CSF analysis is described in
Additional labs performed at that time included B2 microglobulin 1.0, angiotensin converting enzyme <5. The patient was placed on intravenous (IV) acyclovir, broad-spectrum antibiotics, and steroids. Clinically, the patient improved dramatically over the next several days with a resolution of his malaise and headaches, as well as his encephalopathy. On examination, he was oriented × 3 and able to follow complex commands. The laboratory tests for signs of autoimmune disease were all negative, as indicated in

A repeat lumbar puncture performed 5 days later revealed slight improvement in CNS inflammation. Repeat testing through PCR, ELISA, and culture for HSV 1 and 2 remained negative. Repeat imaging showed unchanged T2 hyperintensities in bilateral temporal lobes with improvement in the enhancement pattern of the left temporal lobe [
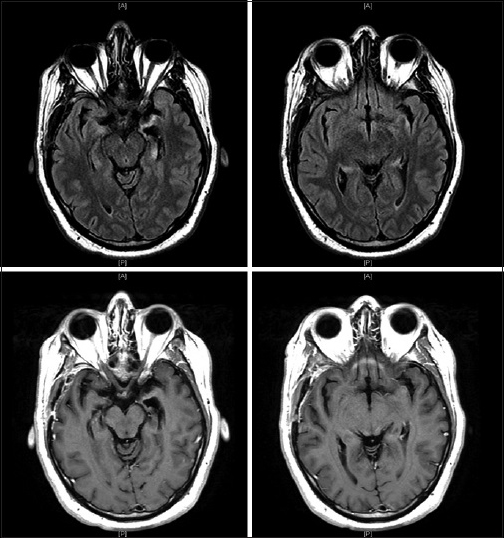
The patient's clinical improvement did not last and approximately 1-week after discharge his short-term memory loss returned and he remained unable to store new memories. He completed his antibiotic course and outpatient follow-up visits over the next 2 months revealed no improvement in his symptomatology. Given the patient's poor quality of life and the absence of diagnosis despite exhaustive testing, the patient was readmitted for right temporal craniotomy and biopsy to establish a tissue diagnosis. Examination at that time was significant for inability to recall items moments after they were told to him. He was awake, alert and oriented to person, place, and time. No focal deficits were noted. A new preoperative MRI showed interval progression of T2 signal abnormality and enhancement of the bilateral temporal lobes with increased mass effect [
The patient tolerated the procedure well. The neurology and rheumatology services saw the patient. He was placed on steroids. Flow cytometry of the biopsy revealed 73% T-cells, 3% mature B-cells with no light chain restriction or significant co-expression of CD5, CD10. Repeat CSF analysis revealed worsening of the inflammatory response and breakdown of the blood-brain barrier [
The final histology revealed transmural and perivascular noncaseating granulomatous vasculitis. Focal vessels showed thickening and hyalinization but were negative for amyloid. No vascular fibrinoid necrosis was seen as would be typical for polyarteritis nodosa, or rheumatoid or lupus vasculitis. Special stains for AFB (Fite), bacteria (gram), and fungus (GMS) were negative.
Immunohistochemical stains for the varicella-zoster virus, Herpes 1 and 2, cytomegalovirus, adenovirus, human herpesvirus 8, EBV, toxoplasma, amyloid were all negative. Repeat testing of autoimmune and infectious etiologies remained negative. Additional studies of paraneoplastic markers were negative including carbohydrate antigen 19–9, CEA, and alpha-FP. Heavy metal screen for lead, arsenic, and mercury all came back below detectable limits. A positron emission tomography scan of the brain showed hypermetabolism of temporal lobes and hippocampal gyri consistent with limbic encephalitis. The patient was discharged with steroids and cyclophosphamide, with persistent anterograde amnesia and a diagnosis of limbic encephalitis secondary to primary CNS vasculitis.
He was treated as an outpatient with IV cyclophosphamide and high dose steroids. Follow-up imaging has shown improvement in both the T2 signal abnormality and contrast enhancement pattern [
DISCUSSION
Primary CNS vasculitis has an estimated incidence rate of 1–2.4 cases per 1 million.[
The clinical manifestations of PCNSV are diverse and nonspecific, and onset can be abrupt, but the course is generally gradual and progressive though in rare cases it can be fulminant.[
Diagnostic laboratory analysis for nonspecific elevated acute phase reactants, ANA, and complement abnormalities, as well as specific markers, such as cytoplasmic anti-neutrophil cytoplasmic antibodies (c-ANCA), perinuclear ANCA (ANCA), and cryoglobulin immune complexes are negative.[
Radiographic analysis with MRI shows abnormalities in the majority of patients. Ischemic changes with diffusion restriction are seen in 50–75% of cases.[
A brain biopsy is required for tissue histopathologic confirmation of PCNSV although the yield is reported at only 50–70% due to the focal segmental nature of the disease.[
Hypophysitis is a rare pituitary inflammatory disorder with an incidence of 1 in 9 million.[
MRI can reveal marked homogeneous enhancement, symmetric enlargement, and dural enhancement.[
Hypophyseal biopsy, in the absence of features of fungus, tuberculosis, and sarcoidosis, is histologically subdivided into three pathologic subtypes: Lymphocytic, granulomatous, and xanthomatous with lymphocytic being the most common.[
Despite the distinction between the two conditions, controversy exists over whether they are truly disparate. The early observation suggested the possibility of a spectrum of progression starting with LYH and progressing to GH.[
Our case was unusual and challenging diagnostically because the initial manifestation of PCNSV was the isolated finding of GH, initially thought to be LYH on frozen section. This uncharacteristically presented in an otherwise previously healthy young male. The clinical course and radiographic appearance of what is now known as the progression of disease initially mimicked herpes encephalitis. A lesson learned is always to consider CNS vasculitis in the differential diagnosis of enhancing CNS lesions in the temporal lobe, particularly when CSF Herpes polymerase chain reaction studies are negative.
The occurrence of GH preceding the onset of primary CNS vasculitis in a previously healthy male is highly unusual and has never before been reported in the literature. Perhaps this suggests an underlying and unifying autoimmune etiology for both GH and PCNSV with a common target found in both hypophyseal and vascular tissue.
Financial support and sponsorship
This work was supported in part by the Donna and Kenneth R. Peak Foundation, The Kenneth R. Peak Brain and Pituitary Center at Houston Methodist Hospital, The Taub Foundation, The Blanche Green Estate Fund of the Pauline Sterne Wolff Memorial Foundation, The Veralan Foundation, The Methodist Hospital Foundation, The American Brain Tumor Association.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
This work was supported in part by the Donna and Kenneth R. Peak Foundation, The Kenneth R. Peak Brain and Pituitary Center at Houston Methodist Hospital, The Taub Foundation, The Blanche Green Estate Fund of the Pauline Sterne Wolff Memorial Foundation, The Veralan Foundation, The Methodist Hospital Foundation, The American Brain Tumor Association, and by many patients and families who have been impacted by the devastating effects of brain tumors and other diseases of the central nervous system.
References
1. Abe T, Matsumoto K, Sanno N, Osamura Y. Lymphocytic hypophysitis: Case report. Neurosurgery. 1995. 36: 1016-9
2. Alrawi A, Trobe JD, Blaivas M, Musch DC. Brain biopsy in primary angiitis of the central nervous system. Neurology. 1999. 53: 858-60
3. Atalay MK, Bluemke DA. Magnetic resonance imaging of large vessel vasculitis. Curr Opin Rheumatol. 2001. 13: 41-7
4. Bellastella A, Bizzarro A, Coronella C, Bellastella G, Sinisi AA, De Bellis A. Lymphocytic hypophysitis: A rare or underestimated disease?. Eur J Endocrinol. 2003. 149: 363-76
5. Beressi N, Beressi JP, Cohen R, Modigliani E. Lymphocytic hypophysitis. A review of 145 cases. Ann Med Interne (Paris). 1999. 150: 327-41
6. Berlit P, Kraemer M. Cerebral vasculitis in adults: What are the steps in order to establish the diagnosis?. Red flags and pitfalls. Clin Exp Immunol. 2014. 175: 419-24
7. Berlit P. Diagnosis and treatment of cerebral vasculitis. Ther Adv Neurol Disord. 2010. 3: 29-42
8. Birnbaum J, Hellmann DB. Primary angiitis of the central nervous system. Arch Neurol. 2009. 66: 704-9
9. Buxton N, Robertson I. Lymphocytic and granulocytic hypophysitis: A single centre experience. Br J Neurosurg. 2001. 15: 242-5
10. Calabrese LH, Dodick DW, Schwedt TJ, Singhal AB. Narrative review: Reversible cerebral vasoconstriction syndromes. Ann Intern Med. 2007. 146: 34-44
11. Calabrese LH, Mallek JA. Primary angiitis of the central nervous system. Report of 8 new cases, review of the literature, and proposal for diagnostic criteria. Medicine (Baltimore). 1988. 67: 20-39
12. Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005. 26: 599-614
13. Cebelin MS, Velasco ME, de las Mulas JM, Druet RL. Galactorrhea associated with lymphocytic adenohypophysitis. Case report. Br J Obstet Gynaecol. 1981. 88: 675-80
14. Folkerth RD, Price DL Jr, Schwartz M, Black PM, De Girolami U. Xanthomatous hypophysitis. Am J Surg Pathol. 1998. 22: 736-41
15. Giannini C, Salvarani C, Hunder G, Brown RD. Primary central nervous system vasculitis: Pathology and mechanisms. Acta Neuropathol. 2012. 123: 759-72
16. Hankey G. Isolated angiitis/angiopathy of the CNS. Prospective diagnostic and therapeutic experience. Cerebrovasc Dis. 1991. 1: 2-15
17. Hashimoto K, Takao T, Makino S. Lymphocytic adenohypophysitis and lymphocytic infundibuloneurohypophysitis. Endocr J. 1997. 44: 1-10
18. Heinze HJ, Bercu BB. Acquired hypophysitis in adolescence. J Pediatr Endocrinol Metab. 1997. 10: 315-21
19. Hunder GG, Salvarani C, Brown RD. Primary central nervous system vasculitis: Is it a single disease?. Ann Neurol. 2010. 68: 573-4
20. Hunn BH, Martin WG, Simpson S, Mclean CA. Idiopathic granulomatous hypophysitis: A systematic review of 82 cases in the literature. Pituitary. 2014. 17: 357-65
21. Imura H, Nakao K, Shimatsu A, Ogawa Y, Sando T, Fujisawa I. Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med. 1993. 329: 683-9
22. Kraemer M, Berlit P. Primary central nervous system vasculitis: Clinical experiences with 21 new European cases. Rheumatol Int. 2011. 31: 463-72
23. Lee JH, Laws ER, Guthrie BL, Dina TS, Nochomovitz LE. Lymphocytic hypophysitis: Occurrence in two men. Neurosurgery. 1994. 34: 159-62
24. Leggett DA, Hill PT, Anderson RJ. ‘stalkitis’ in a pregnant 32-year-old woman: A rare cause of diabetes insipidus. Australas Radiol. 1999. 43: 104-7
25. Ludmerer K, Kissane J. Primary hypothyroidism and hypopituitarism in a young woman. Am J Med. 1984. 77: 319-
26. Lury KM. Inflammatory and infectious processes involving the pituitary gland. Top Magn Reson Imaging. 2005. 16: 301-6
27. Mandell DM, Matouk CC, Farb RI, Krings T, Agid R, terBrugge K. Vessel wall MRI to differentiate between reversible cerebral vasoconstriction syndrome and central nervous system vasculitis: Preliminary results. Stroke. 2012. 43: 860-2
28. McCutcheon IE, Oldfield EH. Lymphocytic adenohypophysitis presenting as infertility. Case report. J Neurosurg. 1991. 74: 821-6
29. Molitch ME, Gillam MP. Lymphocytic hypophysitis. Horm Res. 2007. 68: 145-50
30. Moore PM. Vasculitis of the central nervous system. Curr Rheumatol Rep. 2000. 2: 376-82
31. Nishioka H, Ito H, Sano T, Ito Y. Two cases of lymphocytic hypophysitis presenting with diabetes insipidus: A variant of lymphocytic infundibulo-neurohypophysitis. Surg Neurol. 1996. 46: 285-90
32. Paja M, Estrada J, Ojeda A, Ramón y Cajal S, García-Uría J, Lucas T. Lymphocytic hypophysitis causing hypopituitarism and diabetes insipidus, and associated with autoimmune thyroiditis, in a non-pregnant woman. Postgrad Med J. 1994. 70: 220-4
33. Parisi JE, Moore PM. The role of biopsy in vasculitis of the central nervous system. Semin Neurol. 1994. 14: 341-8
34. Pestell RG, Best JD, Alford FP. Lymphocytic hypophysitis. The clinical spectrum of the disorder and evidence for an autoimmune pathogenesis. Clin Endocrinol (Oxf). 1990. 33: 457-66
35. Portocarrero CJ, Robinson AG, Taylor AL, Klein I. Lymphoid hypophysitis. An unusual cause of hyperprolactinemia and enlarged sella turcica. JAMA. 1981. 246: 1811-2
36. Rivera JA. Lymphocytic hypophysitis: Disease spectrum and approach to diagnosis and therapy. Pituitary. 2006. 9: 35-45
37. Rumana M, Kirmani A, Khursheed N, Besina S, Khalil M. Lymphocytic hypophysitis with normal pituitary function mimicking a pituitary adenoma: A case report and review of literature. Clin Neuropathol. 2010. 29: 26-31
38. Saiwai S, Inoue Y, Ishihara T, Matsumoto S, Nemoto Y, Tashiro T. Lymphocytic adenohypophysitis: Skull radiographs and MRI. Neuroradiology. 1998. 40: 114-20
39. Salvarani C, Brown RD, Calamia KT, Christianson TJ, Huston J, Meschia JF. Primary CNS vasculitis with spinal cord involvement. Neurology. 2008. 70: 2394-400
40. Salvarani C, Brown RD, Calamia KT, Christianson TJ, Weigand SD, Miller DV. Primary central nervous system vasculitis: Analysis of 101 patients. Ann Neurol. 2007. 62: 442-51
41. Salvarani C, Brown RD, Morris JM, Huston J, Hunder GG. Catastrophic primary central nervous system vasculitis. Clin Exp Rheumatol. 2014. 32: S3-4
42. Sautner D, Saeger W, Lüdecke DK, Jansen V, Puchner MJ. Hypophysitis in surgical and autoptical specimens. Acta Neuropathol. 1995. 90: 637-44
43. Scolding N. Can diffusion-weighted imaging improve the diagnosis of CNS vasculitis?. Nat Clin Pract Neurol. 2007. 3: 608-9
44. Scolding NJ, Jayne DR, Zajicek JP, Meyer PA, Wraight EP, Lockwood CM. Cerebral vasculitis – Recognition, diagnosis and management. QJM. 1997. 90: 61-73
45. Scolding NJ. Central nervous system vasculitis. Semin Immunopathol. 2009. 31: 527-36
46. Tanei T, Nakahara N, Takebayashi S, Ito M, Hashizume Y, Wakabayashi T. Primary angiitis of the central nervous system mimicking tumor-like lesion – case report. Neurol Med Chir (Tokyo). 2011. 51: 56-9
47. Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H. Spectrum of different types of hypophysitis: A clinicopathologic study of hypophysitis in 31 cases. Endocr Pathol. 2002. 13: 183-95
48. Thodou E, Asa SL, Kontogeorgos G, Kovacs K, Horvath E, Ezzat S. Clinical case seminar: Lymphocytic hypophysitis: Clinicopathological findings. J Clin Endocrinol Metab. 1995. 80: 2302-11
49. Zuccoli G, Pipitone N, Haldipur A, Brown RD Jr, Hunder G, Salvarani C. Imaging findings in primary central nervous system vasculitis. Clin Exp Rheumatol. 2011. 29: S104-9

