

- Department of Surgery, Neurosurgery Section, King Abdulaziz Medical City, Ministry of National Guard, Jeddah, Saudi Arabia
- College of Medicine, King Saud bin Abdulaziz University for Health Sciences, Jeddah, Saudi Arabia,
- Neurosurgery Unit, Department of Surgical, King Abdulaziz Specialist Hospital, Taif, Saudi Arabia,
- Department of Neurosurgery, King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia.
Correspondence Address:
Fahad Mahmood Okal, Department of Surgery, Neurosurgery Section, King Abdulaziz Medical City, Ministry of National Guard, Jeddah, Saudi Arabia.
DOI:10.25259/SNI_185_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Fahad Mahmood Okal1, Abdulaziz Hamzah2, Adnan Boubaker3, Mohammed H. Aref4. Primary spinal epidural rhabdomyosarcoma: A case report. 24-Mar-2023;14:99
How to cite this URL: Fahad Mahmood Okal1, Abdulaziz Hamzah2, Adnan Boubaker3, Mohammed H. Aref4. Primary spinal epidural rhabdomyosarcoma: A case report. 24-Mar-2023;14:99. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=12214
Date of Submission
24-Feb-2023
Date of Acceptance
08-Mar-2023
Date of Web Publication
24-Mar-2023
Abstract
Background: Rhabdomyosarcoma (RMS) is a malignant childhood tumor that most commonly involves the skeletal muscles of the head and neck, genitourinary tract, limbs, and, rarely, the spine.
Case Description: A 19-year-old male presented with cauda equina symptoms. Magnetic resonance imaging showed a C7/T1 homogeneously enhancing lesion, causing pathological fracture of the T1. Similar lesions were found on T3 and S1-S2 levels. CT-guided biopsy and immunohistochemistry confirmed the diagnosis of highly malignant alveolar RMS. He underwent multi-level laminectomies with partial tumor debulking but was paraplegic postoperatively.
Conclusion: Spinal RMS rarely involves the soft tissues of the spine and should be surgically resected if feasible. Nevertheless, the long-term prognosis is poor regarding tumor recurrence and metastases.
Keywords: Chemotherapy, Neurosurgery, Radiotherapy, Rhabdomyosarcoma, Spine
INTRODUCTION
Rhabdomyosarcoma (RMS) is a malignant tumor originating from mesenchymal cells that commonly occur in the head and neck region, followed by the genitourinary tract and extremities but rarely the spine of children. It is the third most common extracranial tumor after Wilms’ tumor and neuroblastoma. It occurs in 4–7 cases/one million children under the age of 15 years/per year. Sixty-five percentages of cases are diagnosed in children before age six, with very few reported in adults.[
CASE PRESENTATION
A 19-year-old male presented with a 1-month onset of numbness/weakness in both hands and progressive low back pain, gait disturbance, and 4 days of urinary incontinence. On examination, he exhibited mild quadriparesis.
Investigations
Magnetic resonance imaging (MRI) studies demonstrated multifocal involvement of the spine, likely due to metastases. The cervical MRI showed a right anterolateral preverbal/vertebral C7/T1 homogeneously enhancing lesion, a pathologic fracture of the T1 body, and epidural compression [
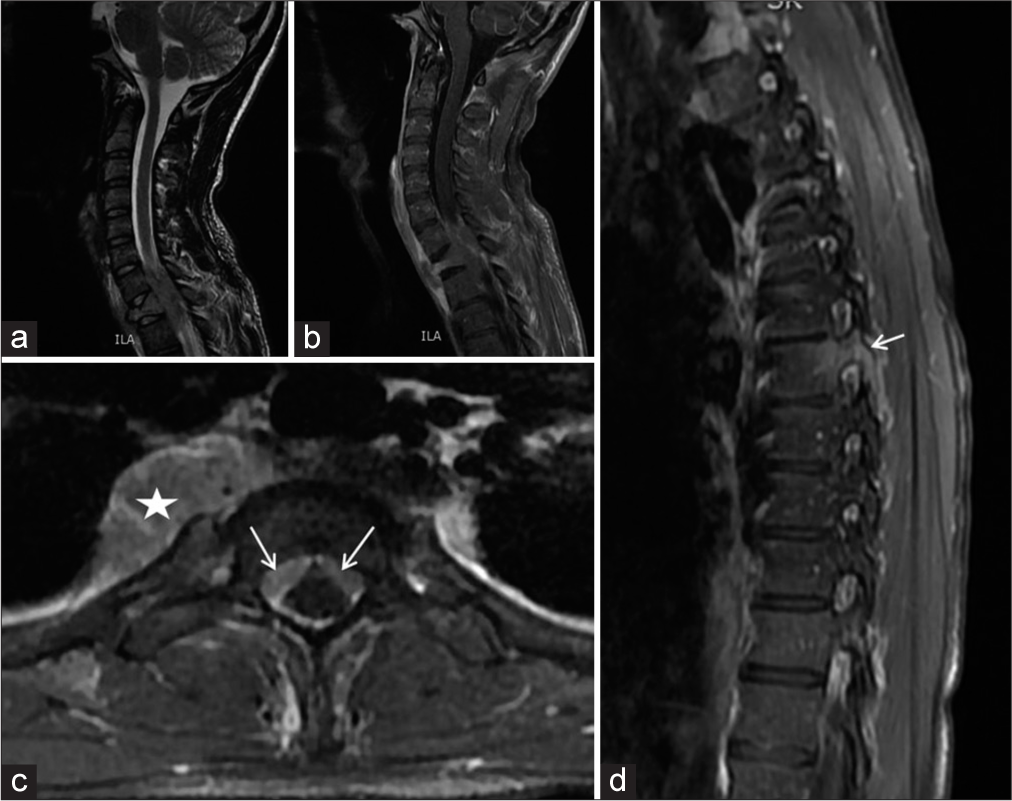
Figure 1:
Cervical and dorsal magnetic resonance imaging in sagittal T2-weighted sequence (a) and T1-weighted sequence with gadolinium (b and d), and in axial T2-weighted sequence at the level of the first dorsal vertebra (c). It shows the pathologic fracture of the latter with the epidural extension of the rhabdomyosarcoma (a and b), also the prevertebral right-sided part of the tumor (star) and its anterior bilateral invasion of the canal causing cord compression (arrows). Note the infiltration of the right pedicle and lower facet of the third dorsal vertebra (d arrow).
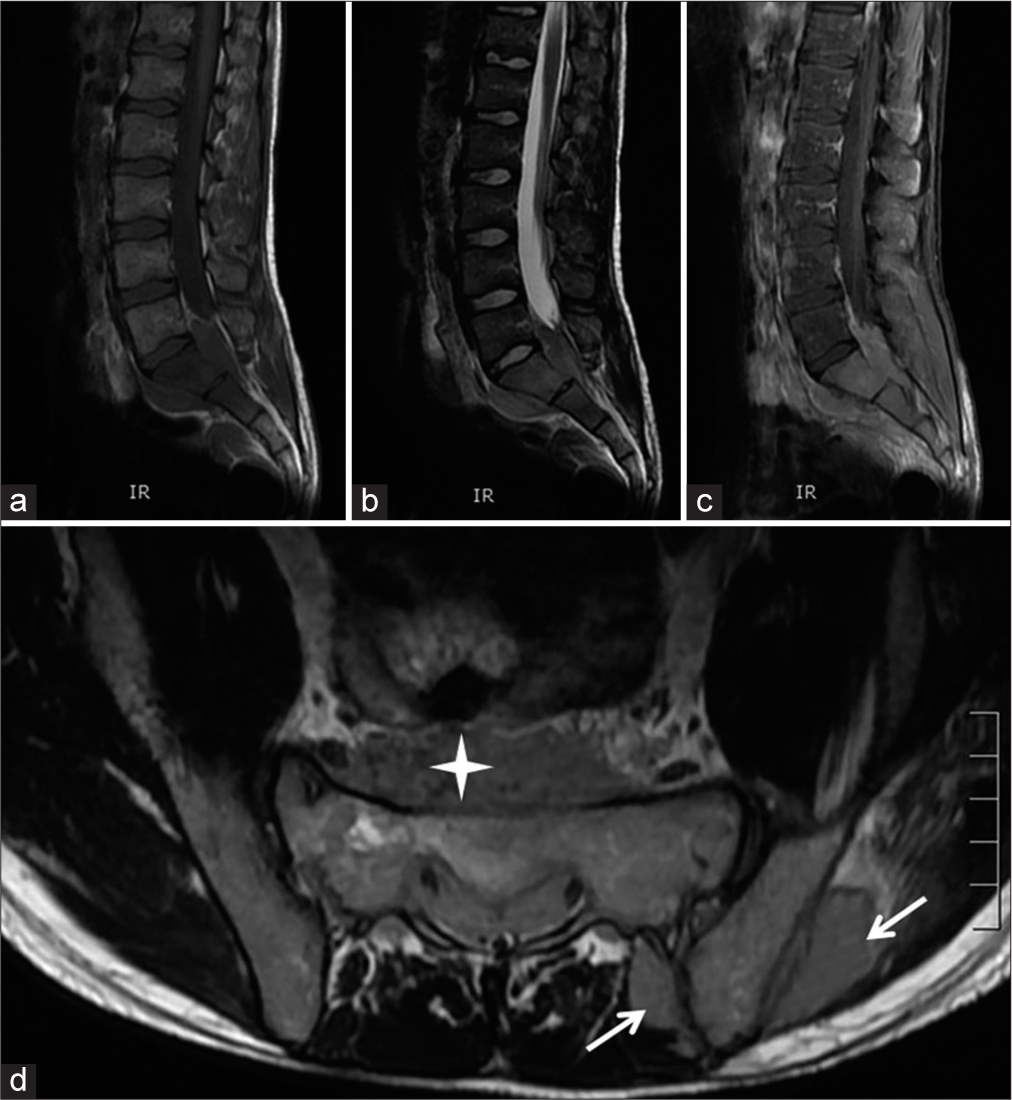
Figure 2:
Lumbosacral magnetic resonance imaging in sagittal cuts T1-weighted sequence (a), T2-weighted sequence (b), and T1 with gadolinium (c), and axial cut T2-weighted sequence, focused on the sacrum. It shows the tumor starting in the pre-sacral space (star), infiltrating the sacrum mainly to the left ala with sacral canal invasion and gluteal muscle infiltration (d arrows).
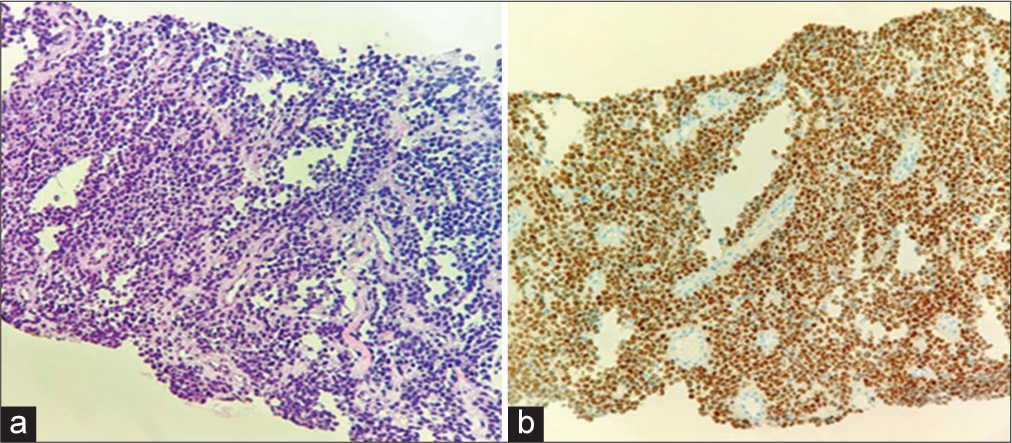
CT-guided biopsy
A CT-guided biopsy of the sacral lesion confirmed the diagnosis. Furthermore, the immunohistochemistry revealed a highly malignant RMS [
Management
Before the initiation of planned chemotherapy/radiation, the patient abruptly developed paraplegia and underwent multilevel decompressive laminectomies for partial debulking of the cervicodorsal and sacral lesions. He remained paraplegic postoperatively and failed to improve over the next 2 months.
DISCUSSION
RMS is the most common soft-tissue sarcoma in the pediatric population, accounting for approximately 5% of all pediatric cancers.[
Radiographic
CT and MRI are the most commonly used imaging modalities in evaluating RMS; however, the findings are variable and non-specific for RMS.[
CT-guided biopsy/pathology
CT-guided biopsy or surgical excision can obtain tissue sampling for the histopathologic examination. According to the current World Health Organization classification, RMS is classified into four subtypes: Embryonal, spindle cell, alveolar, and pleomorphic. The embryonal type is the most common in primary RMS. However, in metastatic RMS, the alveolar subtype is typically found. The alveolar type represents 20% of RMS cases and is associated with a poorer prognosis.[
Management
Treatment typically involves a combination of surgery, radiation therapy, and chemotherapy. At present, there is no consensus on the most effective treatment. Few studies have documented the efficacy of surgery in managing spinal RMS. Wang et al. evaluated 11 primary and secondary RMS cases with posterior locations (eight of the 11 cases); complete surgical resection and spinal fixation with adjuvant radiotherapy provided a longer median overall survival versus palliative surgery/conservative treatment. Therefore, surgery is recommended to treat spinal RMS to reduce the neoplastic mass and allow for a better response to radiation therapy and chemotherapy.[
Radiation therapy is indicated after surgery to eliminate all residual tumor cells and reduce the risk of local recurrence. Chemotherapy is typically given before or after surgery or in combination with radiation therapy.[
CONCLUSION
RMS is a malignant tumor that is exceedingly rare in the spine. Here, a 19-year-old male with primary multifocal/metastatic spinal RMS (C7/T1, T3, and S1/S2) developed paraplegia before radiation and/or chemotherapy could be instituted.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Breneman JC, Lyden E, Pappo AS, Link MP, Anderson JR, Parham DM. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma-a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003. 21: 78-84
2. Dagher R, Helman L. Rhabdomyosarcoma: An overview. Oncologist. 1999. 4: 34-44
3. Khalatbari MR, Jalaeikhoo H, Hamidi M, Moharamzad Y. Primary spinal epidural rhabdomyosarcoma: A case report and review of the literature. Childs Nerv Syst. 2012. 28: 1977-80
4. Kramer S, Meadows AT, Jarrett P, Evans AE. Incidence of childhood cancer: Experience of a decade in a population-based registry. J Natl Cancer Inst. 1983. 70: 49-55
5. Shouman T, El-Kest I, Zaza K, Ezzat M, William H, Ezzat I. Rhabdomyosarcoma in childhood: A retrospective analysis of 190 patients treated at a single institution. J Egypt Natl Cancer Inst. 2005. 17: 67-75
6. Spalteholz M, Gulow J. Pleomorphic rhabdomyosarcoma infiltrating thoracic spine in a 59-year-old female patient: Case report. GMS Interdiscip Plastic Reconstr Surg. 2017. 6: Doc11
7. Van Rijn RR, Wilde JC, Bras J, Oldenburger F, McHugh KM, Merks JH. Imaging findings in noncraniofacial childhood rhabdomyosarcoma. Pediatr Radiol. 2008. 38: 617-34
8. Wang T, Gao X, Yang J, Guo W, Wu Z, Tang L. Treatment strategies and outcomes for spinal rhabdomyosarcoma: A series of 11 cases in a single center and review of the literature. Clin Neurol Neurosurg. 2020. 192: 105729
9. Young JL, Ries LG, Silverberg E, Horm JW, Miller RW. Cancer incidence, survival, and mortality for children younger than age 15 years. Cancer. 1986. 58: 598-602

