

- Department of Neurosurgery, Baylor College of Medicine, Houston, United States
- Department of Neurosurgery, Baylor College of Medicine/Texas Children’s Hospital, Houston, United States
- Department of Pediatric Hematology-Oncology, Baylor College of Medicine, Texas Children’s Cancer Center, Dan L Duncan Cancer Center, Houston, United States
- Department of Pathology, Baylor College of Medicine/Texas Children’s Hospital, Houston, United States.
Correspondence Address:
David Bauer, Department of Neurosurgery, Baylor College of Medicine/Texas Children’s Hospital, Houston, United States.
DOI:10.25259/SNI_825_2022
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Huy Dang1, Abdul Basit Khan1, Nisha Gadgil2, Marc Prablek1, Frank Y. Lin3, Melissa M. Blessing4, Guillermo Aldave2, David Bauer2. Primary spinal intramedullary anaplastic ganglioglioma in a pediatric patient. 17-Feb-2023;14:55
How to cite this URL: Huy Dang1, Abdul Basit Khan1, Nisha Gadgil2, Marc Prablek1, Frank Y. Lin3, Melissa M. Blessing4, Guillermo Aldave2, David Bauer2. Primary spinal intramedullary anaplastic ganglioglioma in a pediatric patient. 17-Feb-2023;14:55. Available from: https://surgicalneurologyint.com/surgicalint-articles/12159/
Date of Submission
07-Sep-2022
Date of Acceptance
31-Jan-2023
Date of Web Publication
17-Feb-2023
Abstract
Background: Gangliogliomas (GGs) are rare tumors of the central nervous system composed of neoplastic neural and glial cells and are typically low-grade. Intramedullary spinal anaplastic GGs (AGG) are rare, poorly understood, and often aggressive tumors that can result in widespread progression along the craniospinal axis. Due to the rarity of these tumors, data are lacking to guide clinical and pathologic diagnosis and standard of care treatment. Here, we present a case of pediatric spinal AGG to provide information on our institutional approach to work-up and to highlight unique molecular pathology.
Case Description: A 13-year-old female presented with signs of spinal cord compression including right sided hyperreflexia, weakness, and enuresis. Magnetic resonance imaging (MRI) revealed a C3-C5 cystic and solid mass which was treated surgically with osteoplastic laminoplasty and tumor resection. Histopathologic diagnosis was consistent with AGG, and molecular testing identified mutations in H3F3A (K27M), TP53, and NF1. She received adjuvant radiation therapy and her neurological symptoms improved. However, at 6-month follow-up, she developed new symptoms. MRI revealed metastatic recurrence of tumor with leptomeningeal and intracranial spread.
Conclusion: Primary spinal AGGs are rare tumors, but a growing body of literature shows some trends that may improve diagnosis and management. These tumors generally present in adolescence and early adulthood with motor/sensory impairment and other spinal cord symptoms. They are most commonly treated by surgical resection but frequently recur due to their aggressive nature. Further reports of these primary spinal AGGs along with characterization of their molecular profile will be important in developing more effective treatments.
Keywords: Anaplastic, Case report, Ganglioglioma, Spinal
INTRODUCTION
Gangliogliomas (GGs) are tumors of the central nervous system (CNS) that arise from glioneuronal precursors. Histologically, they are composed of both neoplastic glial and neural components, with the glial component proliferating and most often driving the tumor pathophysiology.[
Intramedullary/spinal GGs are most often found in the cervical and thoracic spine with symptoms associated with neuroaxis dysfunction in sensation, motor control, and pain.[
CASE DESCRIPTION
History and presentation
A developmentally normal 13-year-old African American female with a history of attention deficit hyperactivity disorder and mild scoliosis presented for evaluation of new onset right elbow contracture. Three years prior, she experienced a sudden inability to straighten her right elbow and simultaneously developed stiffness and pain in the right shoulder. Acutely, she reported recurring episodes of enuresis, concerning for spinal cord pathology. She has no significant family history pertaining to presentation. Physical examination revealed hyperreflexia of her right upper (3 +) and lower extremity (4 + with clonus and positive Babinski reflex). She had right upper and lower extremity weakness (4/5 strength), decreased bulk, and increased tone. No evidence of dysmetria, ataxia, or changes in gait. Magnetic resonance imaging (MRI) of the cervical spine revealed a 4.1 × 1.2 × 1.5 cm mixed cystic and solid intramedullary mass spanning C3-C6 with cord edema extending superiorly and inferiorly to these levels [
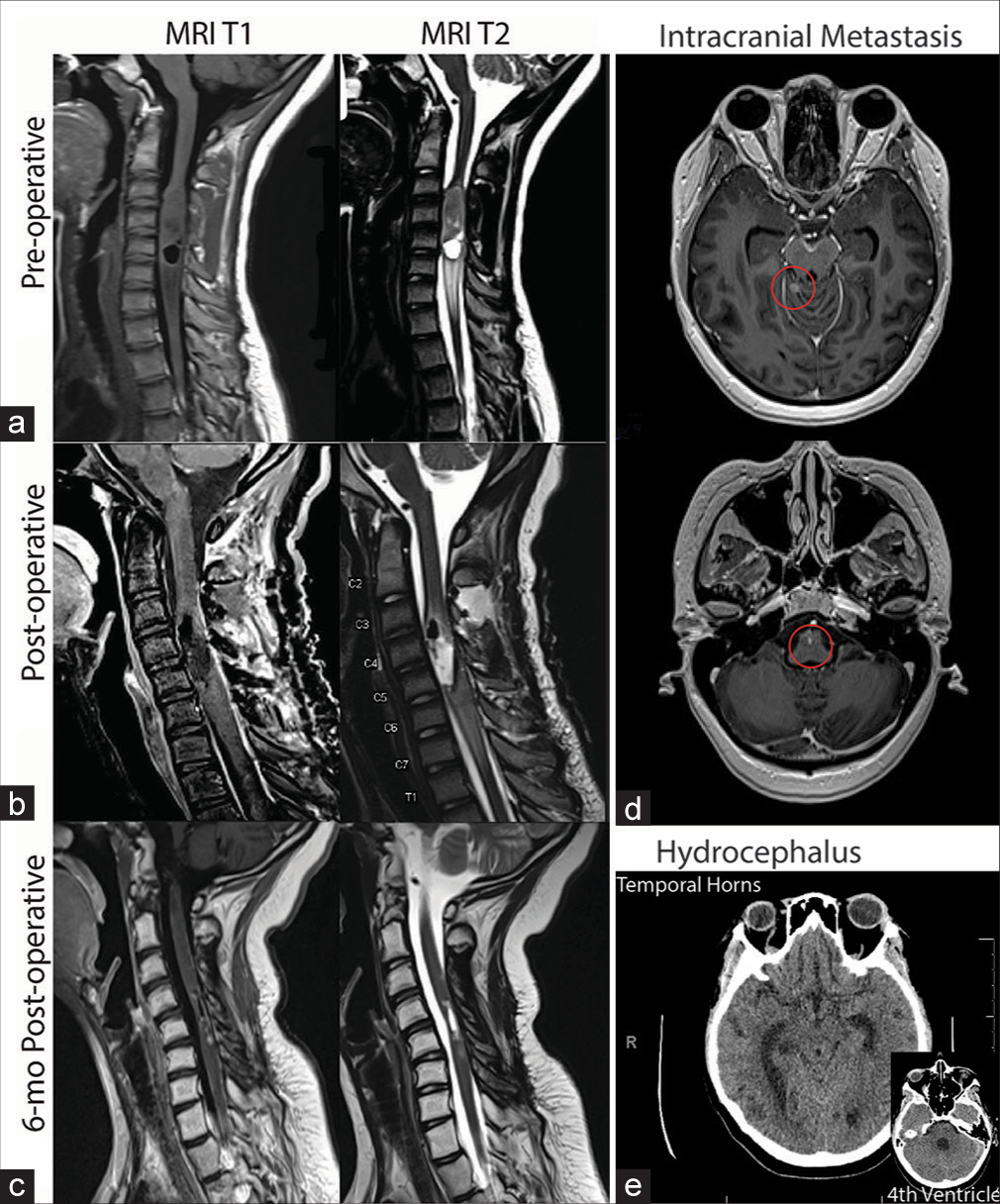
Figure 1:
Magnetic resonance imaging T1 and T2 sequences at (a) preoperative, demonstrating cord expansion suspicious for the presence of a tumor; (b) postoperative, demonstrating gross total resection and subtle enhancement along the posterior aspect of the cord, and (c) 6-month post-operative, demonstrating new leptomeningeal enhancement concerning for metastatic spread of disease. (d) 8-month postoperative (T1 with contrast) demonstrating intracranial metastasis (circled in red) in the cerebellum and brainstem. (e) 8-month postoperative (T1) demonstrating hydrocephalus in the temporal horns of the lateral ventricles and the fourth ventricle.
Surgical procedure
Cervical C3-6 laminectomy was performed followed by semi-laminectomy of inferior C2 and superior C7. A key technical nuance is to perform adequate bony exposure to afford a generous operative window for the tumor. Next, epidural ultrasound was used to identify the tumor, which appeared mixed density but generally hyperechoic. The dura was opened and then direct stimulation was used for dorsal column mapping. Physiologic midline was identified with stimulation, which corresponded to an anatomic midline. After midline myelotomy, we exposed tumor which appeared gray and firm. We exposed the rostral and caudal extent of the tumor. The tumor was biopsied, and then microsurgical technique with an ultrasonic aspirator was used to debulk and resect the tumor. Gross total resection (GTR) of the tumor was achieved without any residual tumor as confirmed by intraoperative ultrasound and visual inspection. Laminoplasty was then performed and previously removed laminae reapproximated. Intraoperative neuromonitoring was used to maximize tumor resection and minimize neurological morbidity.[
Postoperative care and follow-up
Immediate postoperative MRI showed GTR of tumor. The patient did have new temporary worsening deficits in strength, endurance, balance, and mobility after surgery. She remained inpatient for approximately 2 months for rehabilitation, making good recovery. She then received adjuvant proton beam radiation therapy to the C2-7 tumor bed at a dose of 50.4 Gy (relative biological effectiveness). The family declined chemotherapy, including a trial of temozolomide or other early phase therapeutic studies, after careful consideration with the clinical team. At 3-month follow-up, she reported that her weakness and muscle deficits were improved with physical therapy, but she had developed new onset of upper back pain that began around the time she started radiotherapy. MRI at 1 month revealed subtle enhancement around the surgical resection site that may indicate postsurgical scar tissue [
Pathologic diagnosis
Histologic analysis showed a pleomorphic neoplasm with atypical glial and dysmorphic neuronal cells, some of which displayed binucleation [
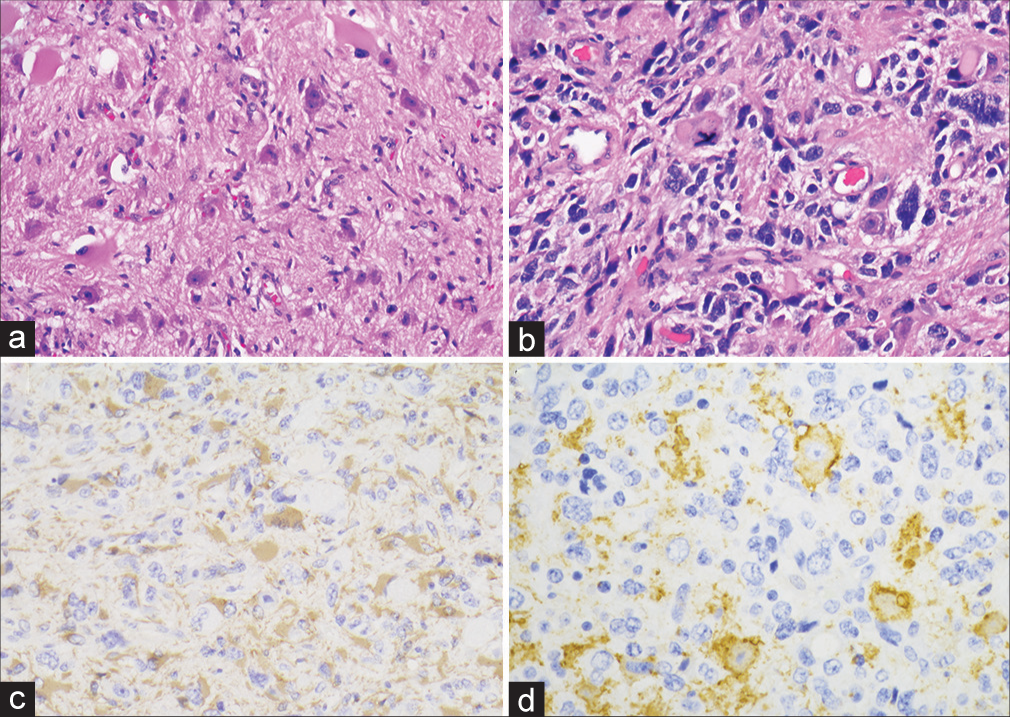
Figure 2:
Histologic sections show a fibrillary background with atypical mature appearing neurons, including binucleate forms (a). Other sections have a higher grade component with marked atypia and atypical mitoses (b). Glial fibrillary acidic protein is positive in the majority of neoplastic cells (c), and Synaptophysin highlights an atypical neuronal component (d).
DISCUSSION
Diagnostic strategies
Intramedullary spinal cord tumors can be difficult to localize and require imaging.[
Histopathologically, GGs are diagnosed by the presence of both glial and neuronal neoplastic cells. Immunohistochemistry including GFAP and synaptophysin, respectively, seem to be the most reliable markers to identify the dual population of neoplastic cells.[
AGGs have been historically diagnosed based on histological criteria. However, diagnostic criteria for AGG continues to be challenging, with attempts to categorize tumors by their molecular profiles.[
Tumor molecular profile
The biologic understanding of pediatric spinal AGGs has been limited by its rare incidence. Alterations involving the MAPK signaling pathway, most frequently BRAF p.V600E mutations, have been reported in a majority of GGs.[
TP53 is commonly mutated in many tumors due to its critical role as a cell cycle checkpoint inhibitor. Cancer cells temper this regulation which allows them to over-proliferate and accumulate more mutations.[
The NF1 mutations found in this tumor were believed to cause premature truncation and loss of function of the NF1 protein based on their location. NF1 loss of function has been observed in many tumor types and results in elevated levels of active GTP-bound RAS and activation of MAPKinase and PI3K pathways.[
Treatments and outcomes
Surgical excision is the standard of care for AGGs with the goal of GTR, but rate of recurrence is high even after complete removal. Adjuvant radiation or chemotherapy is often recommended, though this is controversial because of the scant evidence for its efficacy.[
CONCLUSION
AGGs located in the spinal cord are rare and not well studied. There are currently only 22 cases reported and minimal information available about the genetic profile of AGGs. Our case documents an aggressive spinal AGG that progressed despite surgical resection and adjuvant radiation therapy. Molecular profiling of this tumor revealed genomic mutations including H3F3A, TP53, and NF1. These mutations represent possible prognostic markers, classifiers, and therapeutic targets for the future investigation. As more cases are added to the literature, we look forward to a more well-informed, data-driven approach to recognizing, diagnosing, classifying, and treating this tumor type.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996. 12: 144-8
2. Gessi M, Dörner E, Dreschmann V, Antonelli M, Waha A, Giangaspero F. Intramedullary gangliogliomas: Histopathologic and molecular features of 25 cases. Hum Pathol. 2016. 49: 107-13
3. Gojo J, Pavelka Z, Zapletalova D, Schmook MT, Mayr L, Madlener S. Personalized treatment of H3K27M-Mutant pediatric diffuse gliomas provides improved therapeutic opportunities. Front Oncol. 2020. 9: 1436
4. Hayashi Y, Iwato M, Hasegawa M, Tachibana O, Von Deimling A, Yamashita J. Malignant transformation of a gangliocytoma/ganglioglioma into a glioblastoma multiforme: A molecular genetic analysis. Case report. J Neurosurg. 2001. 95: 138-42
5. Houten JK, Weiner HL. Pediatric intramedullary spinal cord tumors: Special considerations. J Neurooncol. 2000. 47: 225-30
6. Jallo GI, Freed D, Epstein FJ. Spinal cord gangliogliomas: A review of 56 patients. J Neurooncol. 2004. 68: 71-7
7. Joyon N, Tauziède-Espariat A, Alentorn A, Giry M, Castel D, Capelle L. K27M mutation in H3F3A in ganglioglioma grade I with spontaneous malignant transformation extends the histopathological spectrum of the histone H3 oncogenic pathway. Neuropathol Appl Neurobiol. 2017. 43: 271-6
8. Kim NR, Wang KC, Bang JS, Choe G, Park Y, Kim SK. Glioblastomatous transformation of ganglioglioma: Case report with reference to molecular genetic and flow cytometric analysis. Pathol Int. 2003. 53: 874-82
9. Kleinschmidt-DeMasters BK, Donson A, Foreman NK, Dorris K. H3 K27M mutation in gangliogliomas can be associated with poor prognosis. Brain Pathol. 2017. 27: 846-50
10. Lippi G, Plebani M. Integrated diagnostics: The future of laboratory medicine?. Biochem Med (Zagreb). 2020. 30: 010501
11. Lisievici A, Augustin GT, Diana P, Alexandru T, Gheorghe LM, Maria S. Grading gangliogliomas: A short case series with clinico-imagistic and immunohistopathological correlations. Maedica (Bucur). 2018. 13: 241-9
12. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021. 23: 1231-51
13. Lucas JT, Huang AJ, Mott RT, Lesser GJ, Tatter SB, Chan MD. Anaplastic ganglioglioma: A report of three cases and review of the literature. J Neurooncol. 2015. 123: 171-7
14. Mallick S, Benson R, Melgandi W, Giridhar P, Rath GK. Impact of surgery, adjuvant treatment, and other prognostic factors in the management of anaplastic ganglioglioma. Childs Nerv Syst. 2018. 34: 1207-13
15. Mantovani F, Collavin L, del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2018. 26: 199-212
16. Niemeyer B, Marchiori E. Anaplastic ganglioglioma involving the entire length of the spinal cord. Eur Neurol. 2018. 79: 125
17. Oppenheimer DC, Johnson MD, Judkins AR. Ganglioglioma of the spinal cord. J Clin Imaging Sci. 2015. 5: 53
18. Pagès M, Beccaria K, Boddaert N, Saffroy R, Besnard A, Castel D. Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol. 2018. 28: 103-11
19. Patel U, Pinto RS, Miller DC, Handler MS, Rorke LB, Epstein FJ. MR of spinal cord ganglioglioma. AJNR Am J Neuroradiol. 1998. 19: 879-87
20. Pekmezci M, Villanueva-Meyer JE, Goode B, van Ziffle J, Onodera C, Grenert JP. The genetic landscape of ganglioglioma. Acta Neuropathol Commun. 2018. 6: 47
21. Pratt D, Natarajan SK, Banda A, Giannini C, Vats P, Koschmann C. Circumscribed/non-diffuse histology confers a better prognosis in H3K27M-mutant gliomas. Acta Neuropathol. 2018. 135: 299-301
22. Reinhardt A, Pfister K, Schrimpf D, Stichel D, Sahm F, Reuss DE. Anaplastic ganglioglioma-a diagnosis comprising several distinct tumour types. Neuropathol Appl Neurobiol. 2022. 48: e12847
23. Selvanathan SK, Hammouche S, Salminen HJ, Jenkinson MD. Outcome and prognostic features in anaplastic ganglioglioma: Analysis of cases from the SEER database. J Neurooncol. 2011. 105: 539-45
24. Terrier LM, Bauchet L, Rigau V, Amelot A, Zouaoui S, Filipiak I. Natural course and prognosis of anaplastic gangliogliomas: A multicenter retrospective study of 43 cases from the French brain tumor database. Neuro Oncol. 2017. 19: 678-88
25. Verla T, Fridley JS, Khan AB, Mayer RR, Omeis I. Neuromonitoring for intramedullary spinal cord tumor surgery. World Neurosurg. 2016. 95: 108-16
26. Vlachos N, Lampros MG, Zigouris A, Voulgaris S, Alexiou GA. Anaplastic gangliogliomas of the spinal cord: A scoping review of the literature. Neurosurg Rev. 2022. 45: 295-304
27. Wang JL, Hong CS, Otero J, Puduvalli VK, Elder JB. Genetic characterization of a multifocal ganglioglioma originating within the spinal cord. World Neurosurg. 2016. 96: e1-608.e4
28. Watson AL, Anderson LK, Greeley AD, Keng VW, Rahrmann EP, Halfond AL. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of schwann cell tumors reduces tumor grade and multiplicity. Oncotarget. 2014. 5: 1502-14
29. WHO Classification of Tumours Editorial Boa, editors. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: International Agency for Research on Cancer; 2021. p.
30. Wolf HK, Müller MB, Spänle M, Zenther J, Schramm J, Wiestler OD. Ganglioglioma: A detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994. 88: 166-73


