

- Department of Neurological Surgery, University of Miami MILLER School of medicine Jackson Memorial Hospital, Miami, Florida, United States,
- Department of Neurosciences, King Faisal Specialist Hospital and Research Center, Tachassoussi, Riyadh, Saudi Arabia.
Correspondence Address:
Imad N Kanaan, Department of Neurosciences, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia.
DOI:10.25259/SNI_382_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Turki Elarjani1, Meshari Rashed Alhuthayl2, Mahammad Dababo2, Imad N Kanaan2. Rathke cleft cyst apoplexy: Hormonal and clinical presentation. 06-Oct-2021;12:504
How to cite this URL: Turki Elarjani1, Meshari Rashed Alhuthayl2, Mahammad Dababo2, Imad N Kanaan2. Rathke cleft cyst apoplexy: Hormonal and clinical presentation. 06-Oct-2021;12:504. Available from: https://surgicalneurologyint.com/surgicalint-articles/11156/
Date of Submission
18-Apr-2021
Date of Acceptance
01-Jul-2021
Date of Web Publication
06-Oct-2021
Abstract
Background: Rathke cleft cyst (RCC) apoplexy is an uncommon type of lesion that is challenging to diagnose without histopathological samples. Very few articles have been published describing the details of RCC apoplexy. We studied a good number of published articles to analyze its demographics, clinical and hormonal presentations, and outcomes.
Methods: A literature review of English language publications about RCC apoplexy or pituitary apoplexy was conducted using Medline and EMBASE search engines. Thirty articles with histological diagnosis of RCC apoplexy were identified, the earliest of which was published in 1990 and the latest in 2019. We combined the findings of these articles with our own case report and then compared the demographics, clinical and hormonal presentations, and outcomes between RCC apoplexy and pituitary adenoma apoplexy.
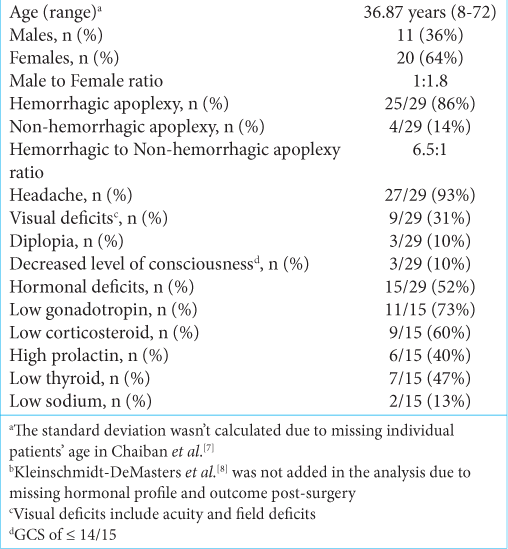
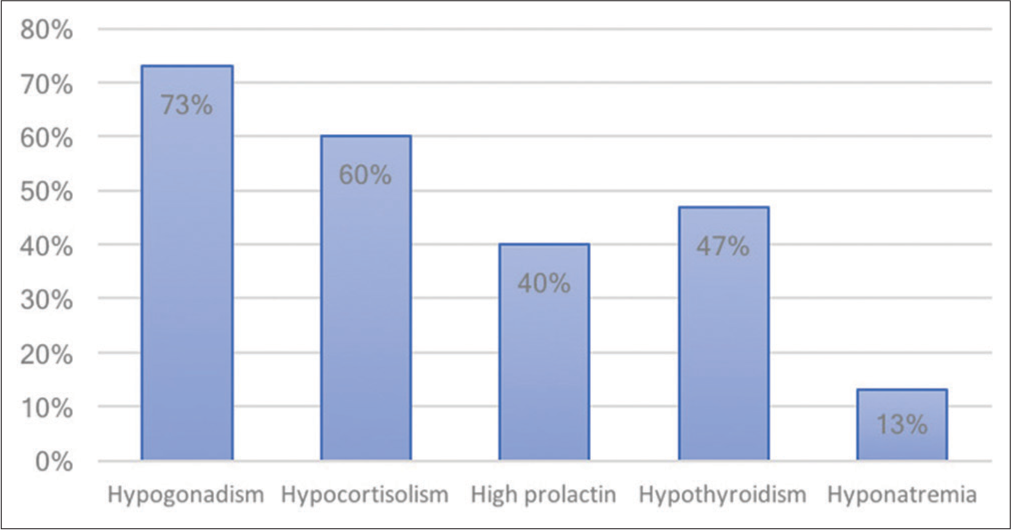
Results: Our data included 29 patients with RCC, with a mean age of 36.87 years (8–72) and a predominance of female patients (68%). The hemorrhagic type was most common, reported in 86%. Headache was the most common presenting symptom, being reported in 93% followed by hypogonadism (73%) and hormonal deficits (52%). All but three patients improved neurologically (90%); however, 45% of patients required long-term hormonal replacement, mostly thyroid hormone. No cases of worsening neurological or hormonal status were reported.
Conclusion: RCC apoplexy presents with less severe neurological and hormonal abnormalities than pituitary adenoma apoplexy; it also has a better prognosis in endocrine functional recovery. We recommend applying current management guidelines of pituitary adenoma apoplexy to RCC apoplexy.
Keywords: Rathke cyst, Apoplexy, Surgery, Pituitary
INTRODUCTION
Rathke cleft cyst (RCC) is an epithelium-lined benign congenital cystic lesion found in the sellar or suprasellar area. RCC originates from the craniopharyngeal duct (Rathke’s pouch).[
MATERIALS AND METHODS
A literature review in Medline, EMBASE, Web of Science, and Scopus was used to identify existing case reports. The search included the following terms: “Rathke’s cleft cyst” AND “Apoplexy” OR “Pituitary apoplexy” OR “Hemorrhagic apoplexy” OR “Nonhemorrhagic apoplexy.” We included all articles with a histological confirmation of RCC apoplexy in any age group, and we excluded non-English articles (two articles). The earliest article found was published in 1990.
The items of interest in each article were patient demographics, clinical presentation, presenting hormonal profile, hemorrhagic versus nonhemorrhagic RCC apoplexy, clinical outcome, and hormonal outcome. Improvement was recorded based on clinical signs and symptoms after management. The database was last searched in February 2020. The article published by Chaiban et al.[
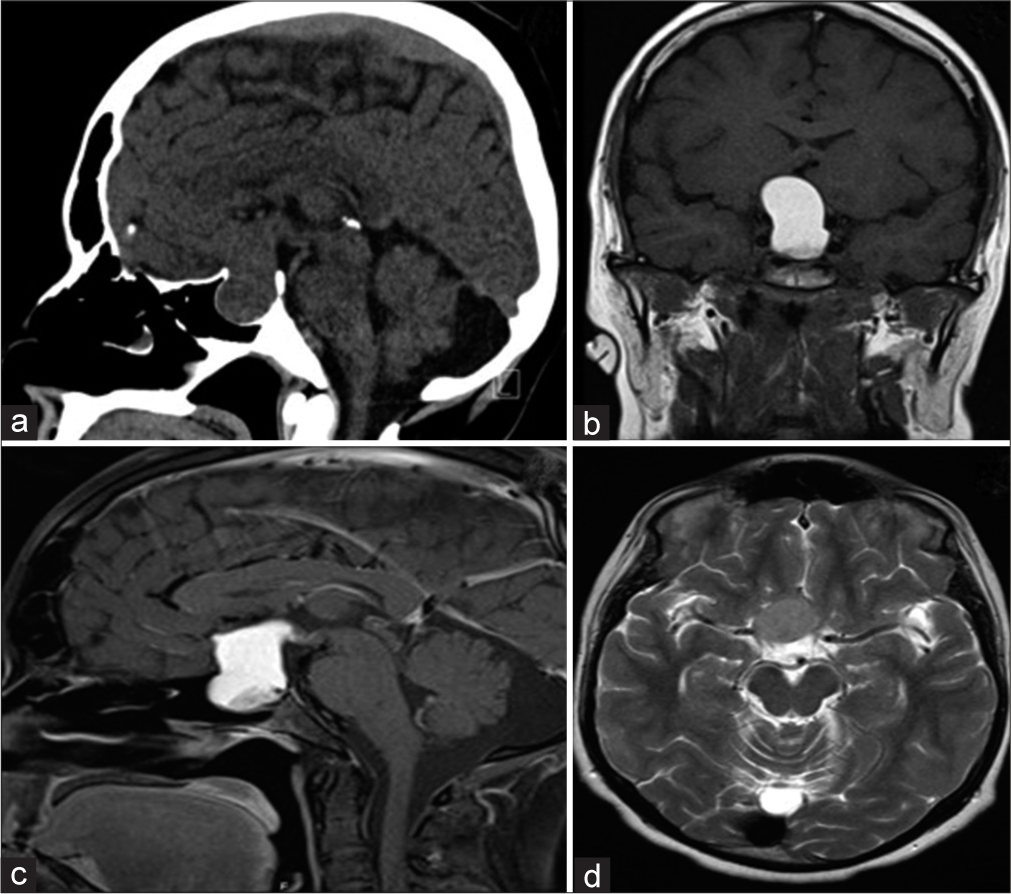
Figure 1:
A 23-year-old man who presented with progressive headache, bitemporal hemianopsia, polydipsia and polyuria, erectile dysfunction, cold intolerance, and constipation for 6 months. Hormonal profile showed low thyroid-stimulating hormone, follicle-stimulating hormone, luteinizing hormone, and testosterone. Sagittal CT scan of the brain demonstrates a large sellar and suprasellar isodense mass with the expansion of the sellar floor (a). Coronal MRI T1 without contrast shows a hyperintense sellar and suprasellar mass compression the third ventricle inferiorly (b). Sagittal MRI T1 with contrast demonstrates a similar hyperintense pattern of the lesion (c). Axial MRI T2 demonstrates an isointense pattern of the lesion and abutting the right clinoidal segment of the internal carotid artery (d).
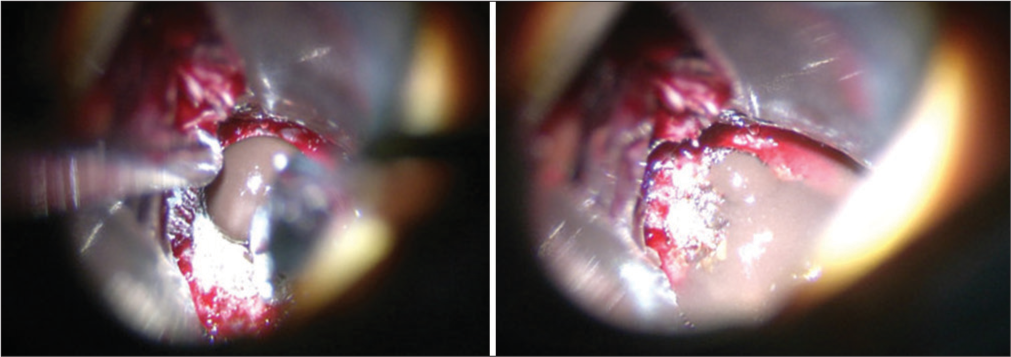
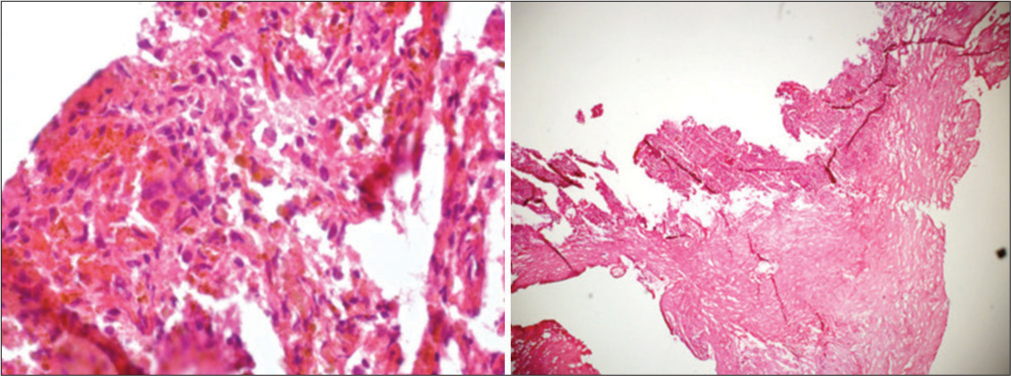
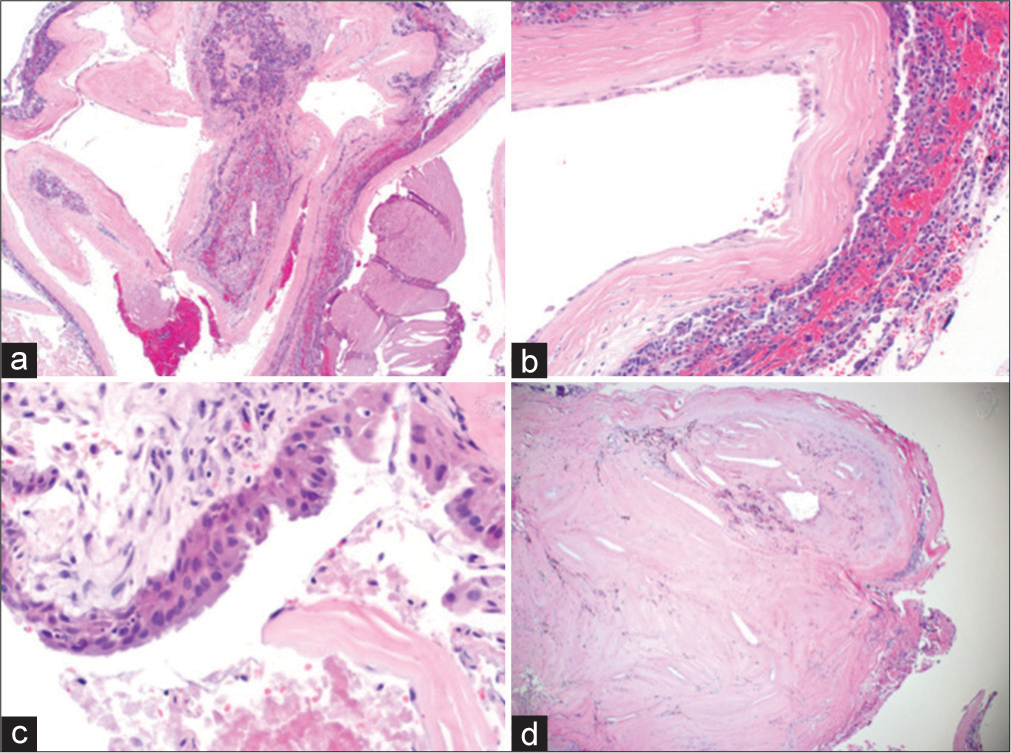
Figure 4:
Permanent section showing cystic lesion with fibrous capsule surrounding nonneoplastic adenohypophysis (a). Higher magnification shows squamous epithelial metaplasia (b). Higher magnification of a different section shows a pseudostratified ciliated columnar epithelium (c). A lower magnification shows fibrosis with cholesterol cleft and old hemorrhage (d).
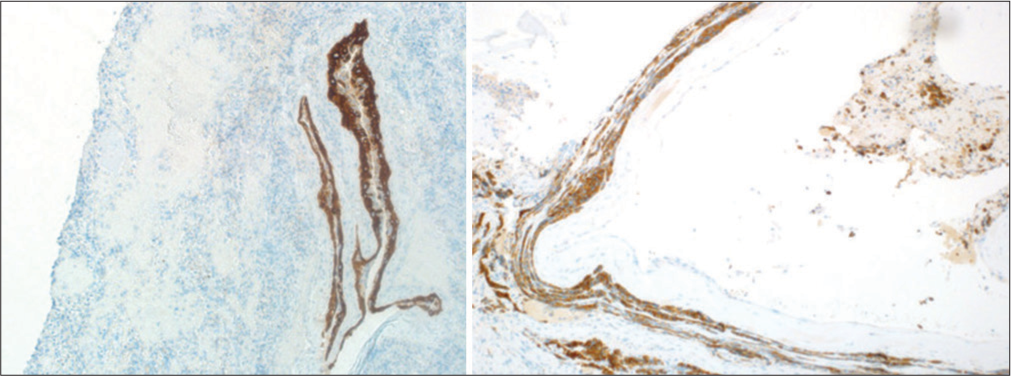
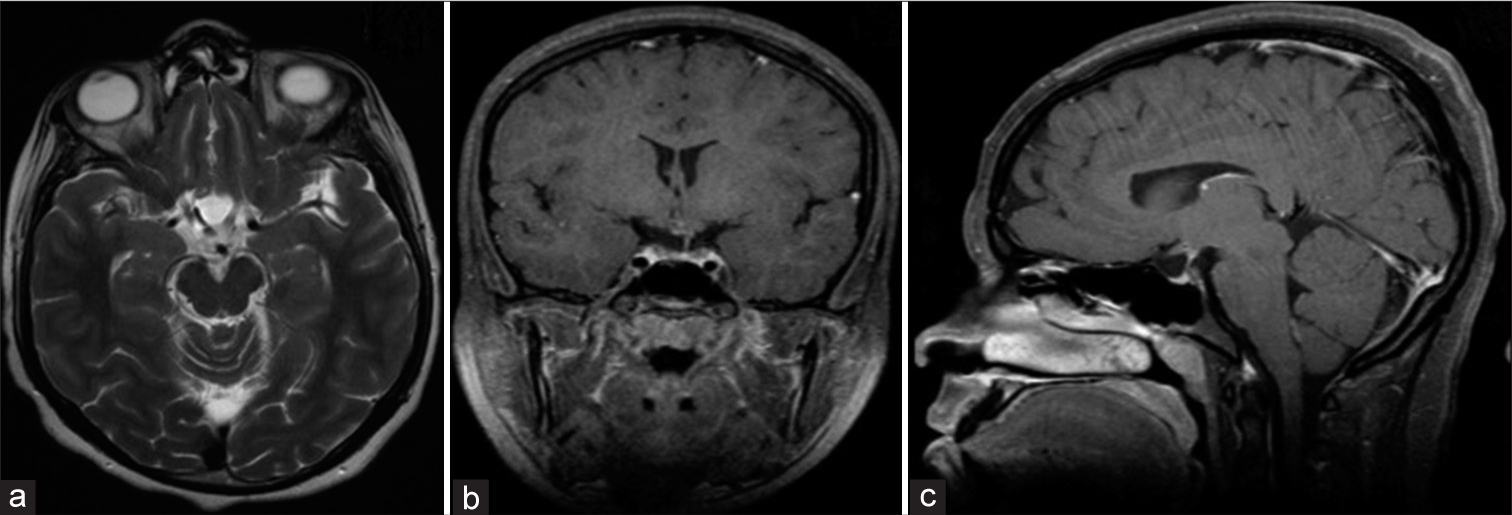
Figure 6:
Postoperative axial MRI T2 (a), coronal T1 without contrast (b), and sagittal T1 with contrast (c) showing decompression of the sella and optic chiasm with drainage of the hemorrhagic cyst. Follow-up after 3 months of surgery demonstrated that the patient has complete improvement of the hormonal profile and visual field.
RESULTS
We identified 29 patients in articles published between 1990 and 2019 [
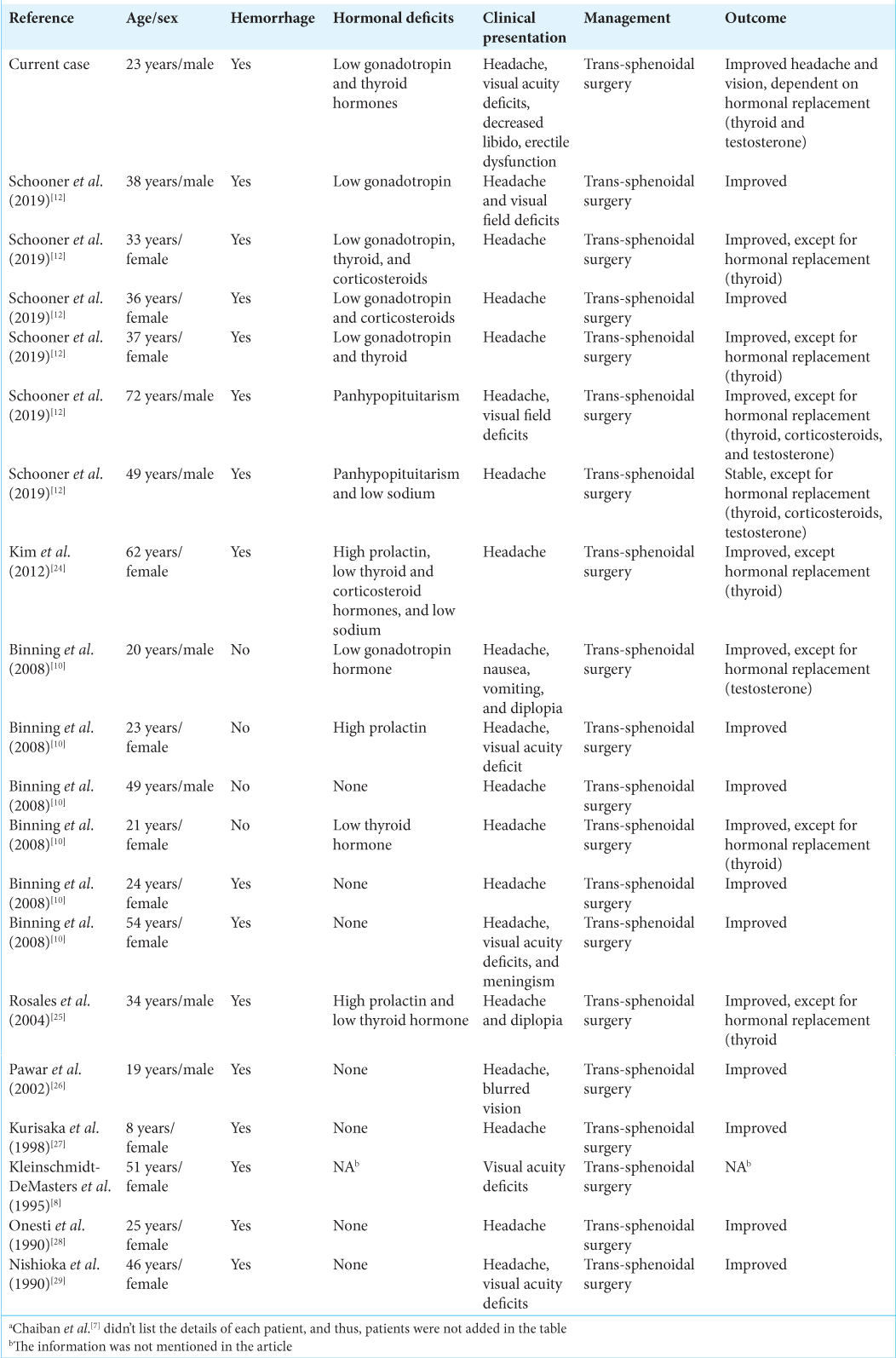
Of the 29 patients in this series, 15 improved without hormonal replacement therapy (52%), 11 improved with continued hormonal replacement therapy (38%), 1 was stable without hormonal replacement therapy (3%), and 2 were stable with continued hormonal replacement therapy (7%) [

DISCUSSION
RCC
RCC is an uncommon cystic lesion that is commonly asymptomatic. The prevalence of RCC among sellar lesions can range from 2% to as high as 7%.[
Its appearance in MRI can be difficult to differentiate from other sellar lesions. Around 70% are hyperintense on T2 with little to no cystic wall enhancement on gadolinium administration.[
RCC apoplexy demographics and pathogenesis
RCC apoplexy is extremely rare, and only a few case reports have been published in the literature. The term RCC apoplexy was coined by Chaiban et al.[
RCC clinical and hormonal presentation
The most common clinical presentation in RCC apoplexy is headache (93%), followed by visual acuity and field deficits (31%), diplopia (10%), and altered level of consciousness (10%). Pituitary adenoma apoplexy has a similar frequency of clinical presentation to RCC apoplexy, with headache being the most common (84–100%), followed by visual field deficits (34–70%), visual acuity deficits (56%), diplopia (45–57%), and decreased level of consciousness (13–30%).[
From the literature review analysis, 52% of RCC apoplexy patients had hormonal abnormalities on presentation, compared to 70–80% of pituitary adenoma apoplexy patients.[
RCC clinical and hormonal outcome
The neurological outcome posttranssphenoidal surgery has improvement in 90% of cases, with 10% having stable neurological deficits. Approximately 45% of patients required long-term hormonal replacement, with 38% improving neurologically. The neurological improvement rate posttranssphenoidal surgery in pituitary adenoma apoplexy is 53–89%; however, 58–83% of patients required long-term hormonal replacement – higher than for RCC apoplexy.[
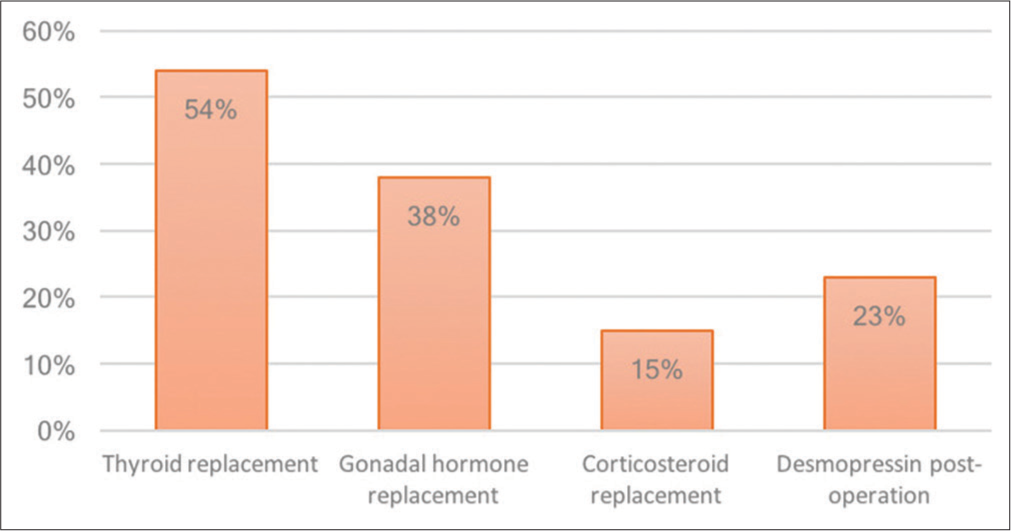
The most common long-term hormonal replacement in RCC apoplexy is thyroid hormone (54%), followed by testosterone replacement (38%), desmopressin for hyponatremia (23%), and cortisol replacement (15%). This is in contrast to pituitary adenoma apoplexy, with testosterone as the most common long-term hormonal replacement (63.6%), followed by thyroid replacement (62.7%), cortisol replacement (60%), and desmopressin (10%). In our case, the patient required long-term thyroid and testosterone replacement. The prevalence of hemorrhagic type RCC apoplexy is 86%, with 85% requiring long-term hormonal replacement.
Treatment guideline
No treatment protocol is designed for RCC apoplexy; however, we can apply the current management plan for pituitary adenoma apoplexy to RCC apoplexy. A review of management guidelines and outcomes of pituitary adenoma apoplexy recommended urgent transsphenoidal microscopic or endoscopic decompression of the RCC lesion for patients presenting with acute or progressive deterioration of their visual field or visual acuity; whereas, other clinical presentations, such as headache, mild decreased level of consciousness, or endocrine abnormalities, were not statistically significant in prompting urgent surgical intervention as compared to conservative management and a wait-and-see approach.[
CONCLUSION
RCC apoplexy is a rare entity that is difficult to diagnose using standard radiological imaging. It has a higher female preponderance and commonly presents in a younger age group in comparison to pituitary adenoma apoplexy. Compared to pituitary adenoma apoplexy, the clinical and hormonal presentation is relatively less severe with a benign postoperative course. Although diabetes insipidus is comparatively more prominent in RCC apoplexy, it is recommended to apply pituitary adenoma apoplexy management guidelines to RCC apoplexy.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abdulbaki A, Kanaan I. The impact of surgical timing on visual outcome in pituitary apoplexy: Literature review and case illustration. Surg Neurol Int. 2017. 8: 16
2. Aho CJ, Liu C, Zelman V, Couldwell WT, Weiss MH. Surgical outcomes in 118 patients with Rathke cleft cysts. J Neurosurg. 2005. 102: 189-93
3. Amhaz HH, Chamoun RB, Waguespack SG, Shah K, McCutcheon IE. Spontaneous involution of Rathke cleft cysts: Is it rare or just underreported?. J Neurosurg. 2010. 112: 1327-32
4. Bi WL, Dunn IF, Laws ER. Pituitary apoplexy. Endocrine. 2015. 48: 69-75
5. Binning MJ, Liu JK, Gannon J, Osborn AG, Couldwell WT. Hemorrhagic and nonhemorrhagic Rathke cleft cysts mimicking pituitary apoplexy. J Neurosurg. 2008. 108: 3-8
6. Briet C, Salenave S, Bonneville JF, Laws ER, Chanson P. Pituitary apoplexy. Endocr Rev. 2015. 36: 622-45
7. Bujawansa S, Thondam SK, Steele C, Cuthbertson DJ, Gilkes CE, Noonan C. Presentation, management and outcomes in acute pituitary apoplexy: A large single-centre experience from the United Kingdom. Clin Endocrinol (Oxf). 2014. 80: 419-24
8. Chaiban JT, Abdelmannan D, Cohen M, Selman WR, Arafah BM. Rathke cleft cyst apoplexy: A newly characterized distinct clinical entity. J Neurosurg. 2011. 114: 318-24
9. Kasperbauer JL, Orvidas LJ, Atkinson JL, Abboud CF. Rathke cleft cyst: Diagnostic and therapeutic considerations. Laryngoscope. 2002. 112: 1836-9
10. Kleinschmidt-DeMasters BK, Lillehei KO, Stears JC. The pathologic, surgical, and MR spectrum of Rathke cleft cysts. Surg Neurol. 1995. 44: 19-26
11. Larkin S, Karavitaki N, Ansorge O. Rathke’s cleft cyst. Handb Clin Neurol. 2014. 124: 255-69
12. Mukherjee JJ, Islam N, Kaltsas G, Lowe DG, Charlesworth M, Afshar F. Clinical, radiological and pathological features of patients with Rathke’s cleft cysts: Tumors that may recur. J Clin Endocrinol Metab. 1997. 82: 2357-62
13. Oka H, Kawano N, Suwa T, Yada K, Kan S, Kameya T. Radiological study of symptomatic Rathke’s cleft cysts. Neurosurgery. 1994. 35: 632-7
14. Ross DA, Norman D, Wilson CB. Radiologic characteristics and results of surgical management of Rathke’s cysts in 43 patients. Neurosurgery. 1992. 30: 173-9
15. Saeger W, Lüdecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S. Pathohistological classification of pituitary tumors: 10 Years of experience with the German pituitary tumor registry. Eur J Endocrinol. 2007. 156: 203-16
16. Schooner L, Wedemeyer MA, Bonney PA, Lin M, Hurth K, Mathew A. Hemorrhagic presentation of Rathke cleft cysts: A surgical case series. Oper Neurosurg (Hagerstown). 2019. 18: 470-9
17. Semple PL, Webb MK, de Villiers JC, Laws ER. Pituitary apoplexy. Neurosurgery. 2005. 56: 65-72
18. Shin JL, Asa SL, Woodhouse LJ, Smyth HS, Ezzat S. Cystic lesions of the pituitary: Clinicopathological features distinguishing craniopharyngioma, Rathke’s cleft cyst, and arachnoid cyst. J Pathol. 1999. 203: 814-21
19. Singh TD, Valizadeh N, Meyer FB, Atkinson JL, Erickson D, Rabinstein AA. Management and outcomes of pituitary apoplexy. J Neurosurg. 2015. 122: 1450-7
20. Teramoto A, Hirakawa K, Sanno N, Osamura Y. Incidental pituitary lesions in 1, 000 unselected autopsy specimens. Radiology. 1994. 193: 161-4
21. Voelker JL, Campbell RL, Muller J. Clinical, radiographic, and pathological features of symptomatic Rathke’s cleft cysts. J Neurosurg. 1991. 74: 535-44
22. Zada G. Rathke cleft cysts: A review of clinical and surgical management. Neurosurg Focus. 2011. 31: E1
23. Zhong W, You C, Jiang S, Huang S, Chen H, Liu J. Symptomatic Rathke cleft cyst. J Clin Neurosci. 2012. 19: 501-8

