

- Division of Neurosurgery, University of Arizona, Tucson, Arizona, United States
- Department of Pathology, University of Arizona, Tucson, Arizona, United States
- Department of Otolaryngology, University of Arizona, Tucson, Arizona, United States
- College of Medicine, University of Arizona, Tucson, Arizona, United States
Correspondence Address:
Nikolay L. Martirosyan
Division of Neurosurgery, University of Arizona, Tucson, Arizona, United States
DOI:10.25259/SNI-83-2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Edvin Telemi, Nikolay L. Martirosyan, Mauricio J. Avila, Ashley L. Lukefahr, Christopher Le, G. Michael Lemole. Suprasellar pleomorphic xanthoastrocytoma: A case report. 24-Apr-2019;10:72
How to cite this URL: Edvin Telemi, Nikolay L. Martirosyan, Mauricio J. Avila, Ashley L. Lukefahr, Christopher Le, G. Michael Lemole. Suprasellar pleomorphic xanthoastrocytoma: A case report. 24-Apr-2019;10:72. Available from: http://surgicalneurologyint.com/surgicalint-articles/9281/
Date of Submission
24-Jul-2018
Date of Acceptance
28-Nov-2018
Date of Web Publication
24-Apr-2019
Abstract
Background:Pleomorphic xanthoastrocytoma (PXA) is a rare form of astrocytic neoplasm most commonly found in children and young adults. This neoplasm, which is classified as a Grade II tumor by the World Health Organization classification of tumors of the central nervous system, carries a relatively favorable outcome. It is usually found supratentorially in cortical regions of the cerebral hemispheres, and as such, presenting symptoms are similar to other supratentorial cortical neoplasms; with seizures being a common initial symptom. Due to the rarity of this type of neoplasm, PXA arising elsewhere in the brain is often not included in the initial differential diagnosis.
Case Description:This report presents an extremely rare patient with PXA arising in the suprasellar region who presented with progressive peripheral vision loss. Magnetic resonance imaging of the brain demonstrated a heterogeneous suprasellar mass with cystic and enhancing components initially; the most likely differential diagnosis was craniopharyngioma. The patient underwent endoscopic endonasal resection of the tumor. Microscopically, the tumor was consistent with a glial neoplasm with variable morphology. Based on these findings along with further immunohistochemical workup, the patient was diagnosed with a PXA arising in the suprasellar region. At the 1-year follow-up, the patient remained free of recurrence. Although rare PXA originating in other uncommon locations, such as the spinal cord, cerebellum, the ventricular system, and the pineal region have been previously described.
Conclusion:Although rare, PXA should be included in the differential diagnosis for solid-cystic tumors arising in the suprasellar region in young adults.
Keywords: Brain neoplasm, endonasal endoscopic approach, optic chiasm lesion, pleomorphic xanthoastrocytoma, suprasellar astrocytoma
INTRODUCTION
Pleomorphic xanthoastrocytoma (PXA), first described by Kepes et al.[
Given their predilection for the temporal lobes, many patients with PXA present with progressive headaches or seizures.[
CASE REPORT
A 30-year-old man with a 3-year history of progressive loss of peripheral vision in his right eye was referred to our clinic following the discovery of a suprasellar mass on imaging. He denied any headaches, history of seizures, recent changes in weight, breast discharge, heat or cold intolerance, or changes in shoe size or facial features. His only complaint was a progressive visual field defect in the right upper quadrant of the right eye. He had no significant other medical or surgical history, and, apart from a visual field defect of the right eye (quadrantanopia), he had an otherwise unremarkable physical exam including intact cranial nerves III to XII. Endocrine workup was unremarkable. Magnetic resonance imaging (MRI) with and without contrast of his brain revealed a multilobulated complex mass with both cystic and solid components in the suprasellar region measuring 28.7 mm × 34.5 mm × 37.2 mm (AP by TR by CC) [
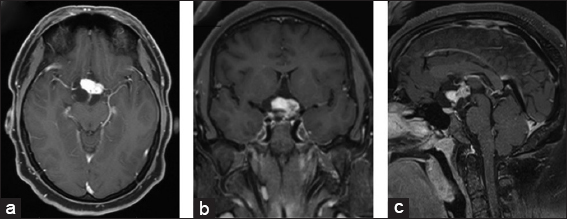
Figure 1
Preoperative magnetic resonance imaging (MRI) of the brain postcontrast T1-weighted MRI (a) axial view, (b) coronal view, (c) sagittal view reveals multilobulated complex mass with both cystic and solid components again seen in the suprasellar region measuring 28.7 mm × 34.5 mm × 37.2 mm (AP by TR by CC).
Operation and postoperative course
The patient underwent surgery for the resection of the suprasellar tumor through an endoscopic endonasal transsphenoidal approach. The intraoperative visualization of the tumor showed that the mass appeared to be arising from the optic chiasm rather than the Sella. Furthermore, intraoperative resection was complicated by significant adherence of the tumor capsule to local structures, including the carotid arteries. To prevent damage to the surrounding structures, the operation was concluded with an estimated 50–60% of the tumor resected. Intraoperative frozen pathology sections results revealed nonspecific glial reactive tissue. Postoperatively, the patient remained at neurologic baseline except for a new left lower quadrant visual field defect. He did not have any oculomotor nerve palsy. MRI confirmed expected partial resection of the suprasellar mass as well as the decreased mass effect on surrounding tissues [
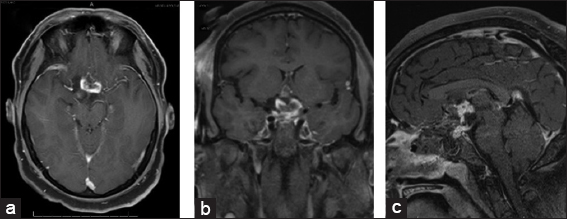
At 1-year follow-up, MRI of his brain revealed the stable size of the known residual with no new areas of enhancement and improvement of the tumor local mass effect. The patient endorsed subjective improvement of his vision, and ophthalmologic follow-up at 1 year revealed partial improvement of his visual fields deficits compared to preoperative evaluation. At 1-year follow-up, the patient was not in any hormonal supplementation.
Pathological analysis
Microscopically, the tumor was consistent with a glial neoplasm with variable morphology. Some cells were spindled, others were within a mucin-rich matrix, and focal cells with lipidized cytoplasm were present. Eosinophilic granular bodies, lymphocytic infiltrates, and Rosenthal fibers were also noted to be present [
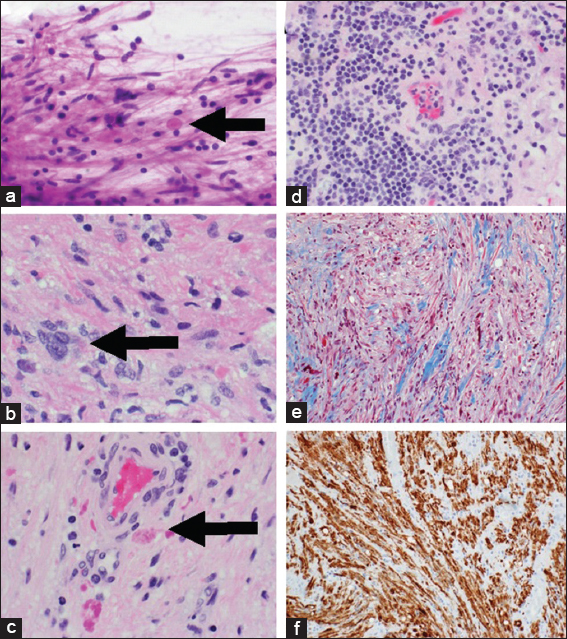
Figure 3
A squash preparation (a) H and E ×40) shows spindled cells with fibrillary astrocytic processes and Rosenthal fibers (arrow).Tumor shows focal cellular atypia with occasional “lipidized” cytoplasm (b) H and E; ×60; arrow), readily identifiable Rosenthal fibers (b and c), and numerous eosinophilic granular bodies (c) H and E; ×60; arrow). Perivascular cuffing, chronic lymphocytic infiltrate is present (d) H and E; ×40). Trichrome stain (e) ×20 highlights the blue-staining connective tissue into which the tumor cells are invading, immunohistochemistry for glial fibrillary acidic protein (f) ×20) highlights the astrocytic processes of the tumor cells.
DISCUSSION
PXA are a rare form of astrocytic tumor that is commonly found in young adults and children.[
As our understanding of this tumor changes, new tests are available to further characterize the tumor histologically. The BRAF and V600E mutation, if present, are associated with longer overall survival compared to the tumors that are nonmutant.[
As mentioned before PXA is usually a supratentorial tumor with a predisposition to the temporal lobes however, PXA has been sporadically reported to arise in unusual locations including the posterior fossa, spinal cord, and the retina.[
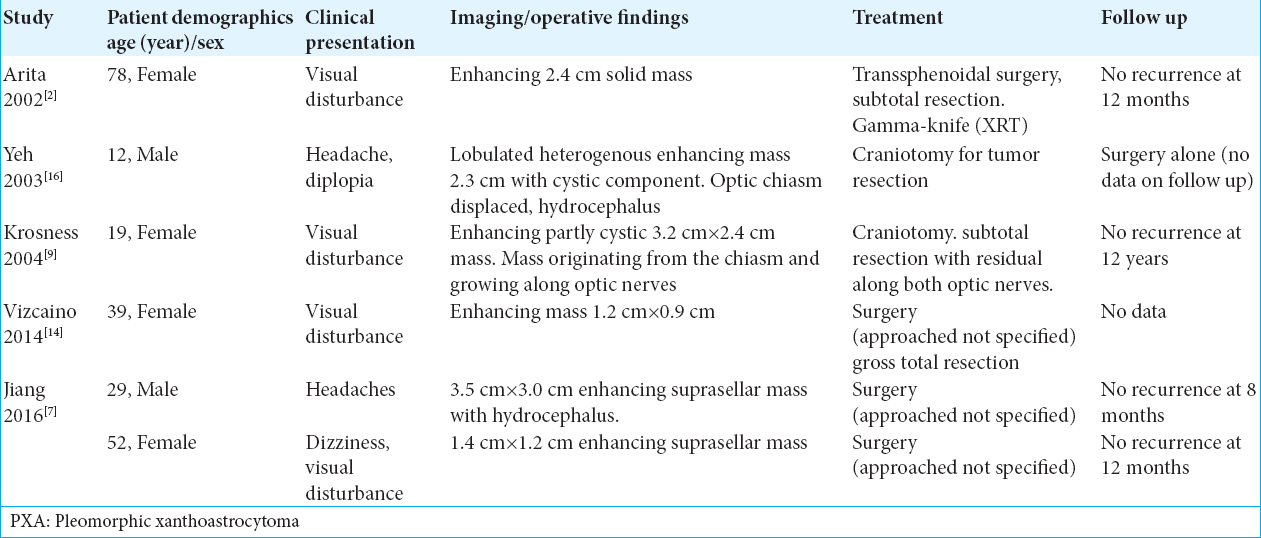
On imaging, PXA often presents as an enhancing, solid-cystic mass on MRI, and commonly causes visual disturbances as the presenting clinical symptom due to local mass effect. Interestingly, as with our case, the case by Yeh et al.[
As mentioned before by having PXA as a differential diagnosis preoperatively, surgical plan may change to have a more aggressive resection to provide a longer recurrence-free survival. In the larger series of PXAs (74 cases), Ida et al.[
At present, there is not a definite guide to treat PXA that did not have a gross total resection during surgery. As mentioned before in general for all PXA, a gross total resection is one of the most important factors for recurrence-free survival.[
CONCLUSION
PXA are rare neoplasms, but they should be considered in the differential diagnosis of suprasellar solid-cystic tumors, especially those found in children and young adults. Gross total resection provides the best overall recurrence free for these tumors and should be the initial goal of surgery: coadjuvant therapy can be considered for tumors with anaplastic features.
Declaration of patient consent
Patient consent was not obtained as the patient is covered under the Institutional Review Boards protocol.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Alexiou GA, Moschovi M, Stefanaki K, Prodromou C, Sfakianos G, Prodromou N. Malignant progression of a pleomorphic xanthoastrocytoma in a child. Neuropediatrics. 2010. 41: 69-71
2. Arita K, Kurisu K, Tominaga A, Sugiyama K, Sumida M, Hirose T. Intrasellar pleomorphic xanthoastrocytoma:Case report. Neurosurgery. 2002. 51: 1079-82
3. Chang HT, Latorre JG, Hahn S, Dubowy R, Schelper RL. Pediatric cerebellar pleomorphic xanthoastrocytoma with anaplastic features:A case of long-term survival after multimodality therapy. Childs Nerv Syst. 2006. 22: 609-13
4. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B. Pleomorphic xanthoastrocytoma:What do we really know about it?. Cancer. 1999. 85: 2033-45
5. Hamlat A, Le Strat A, Guegan Y, Ben-Hassel M, Saikali S. Cerebellar pleomorphic xanthoastrocytoma:Case report and literature review. Surg Neurol. 2007. 68: 89-94
6. Ida CM, Rodriguez FJ, Burger PC, Caron AA, Jenkins SM, Spears GM. Pleomorphic xanthoastrocytoma:Natural history and long-term follow-up. Brain Pathol. 2015. 25: 575-86
7. Jiang GY, Yu JH, Zhang XY, Qi XL, Sun YS. Pleomorphic xanthoastrocytoma arising from the suprasellar region:A report of two cases. J Clin Neurosci. 2016. 33: 228-31
8. Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma:A distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979. 44: 1839-52
9. Krossnes BK, Mella O, Wester K, Mørk SJ. Pigmented astrocytoma with suprasellar location:Case report and literature review. Acta Neuropathol. 2004. 108: 461-6
10. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007. 114: 97-109
11. Nakamura M, Chiba K, Matsumoto M, Ikeda E, Toyama Y. Pleomorphic xanthoastrocytoma of the spinal cord. Case report. J Neurosurg Spine. 2006. 5: 72-5
12. Rippe DJ, Boyko OB, Radi M, Worth R, Fuller GN. MRI of temporal lobe pleomorphic xanthoastrocytoma. J Comput Assist Tomogr. 1992. 16: 856-9
13. Sharma A, Nand Sharma D, Kumar Julka P, Kishor Rath G. Pleomorphic xanthoastrocytoma a clinico-pathological review. Neurol Neurochir Pol. 2011. 45: 379-86
14. Vizcaíno MA, Caccamo DV, Fox E, Rodriguez FJ. Pleomorphic xanthoastrocytoma:Report of two cases with unconventional clinical presentations. Clin Neuropathol. 2014. 33: 380-7
15. Walker M. Diagnostic Pathology Neuropathology. Neuropathology and Applied Neurobiology. 2014. 40: 664-6
16. Yeh DJ, Hessler RB, Stevens EA, Lee MR. Composite pleomorphic xanthoastrocytoma-ganglioglioma presenting as a suprasellar mass:Case report. Neurosurgery. 2003. 52: 1465-8
17. Zarate JO, Sampaolesi R. Pleomorphic xanthoastrocytoma of the retina. Am J Surg Pathol. 1999. 23: 79-81


Javier Hortal
Posted April 28, 2019, 3:21 am
Good morning. My name is Javier Hortal and I live in Spain.
I am the father of a boy who died in 2016. His tumor was a grade 3 xanthoastrocytoma.
A few years ago I decided to create a blog about xanthoastrocytoma to give information and contact all those who had contact with the tumor.
Any information that we can share will be welcome to show and promote it.
This blog wants to be a window open to all information, research, news, experiences and opinions that exist on xanthoastrocytoma. It is not scientific but it is informative. It has three objectives: – Share information. – Promote research. – Connect amongst themselves those interested in this tumor: patients, relatives, doctors and information and communication media
Sincerely.
Javier Hortal