

- Department of Orthopaedics and Rehabilitation, Yale University School of Medicine, New Haven, CT, USA
- Department of Neurosurgery, NYU School of Medicine, Brooklyn, New York, USA
- Department of Radiation Oncology, NYU School of Medicine, Brooklyn, New York, USA
- Department of Radiology, NYU School of Medicine, Brooklyn, New York, USA
- Kimmel Center for Stem Cell Biology, NYU School of Medicine, Brooklyn, New York, USA
- Brain Tumor Center, NYU School of Medicine, Brooklyn, New York, USA
- Division of Neurosurgery, Maimonides Medical Center, Brooklyn, New York, USA
- Maimonides Cancer Center, Brooklyn, New York, USA
Correspondence Address:
Dimitris G Placantonakis
Department of Neurosurgery, NYU School of Medicine, Brooklyn, New York, USA
Kimmel Center for Stem Cell Biology, NYU School of Medicine, Brooklyn, New York, USA
Brain Tumor Center, NYU School of Medicine, Brooklyn, New York, USA
DOI:10.4103/2152-7806.189296
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Tyagi V, Theobald J, Barger J, Bustoros M, Bayin NS, Modrek AS, Kader M, Anderer EG, Donahue B, Fatterpekar G, Placantonakis DG. Traumatic brain injury and subsequent glioblastoma development: Review of the literature and case reports. Surg Neurol Int 26-Aug-2016;7:78
How to cite this URL: Tyagi V, Theobald J, Barger J, Bustoros M, Bayin NS, Modrek AS, Kader M, Anderer EG, Donahue B, Fatterpekar G, Placantonakis DG. Traumatic brain injury and subsequent glioblastoma development: Review of the literature and case reports. Surg Neurol Int 26-Aug-2016;7:78. Available from: http://surgicalneurologyint.com/surgicalint_articles/traumatic-brain-injury-subsequent-glioblastoma-development-review-literature-case-reports/
Abstract
Background:Previous reports have proposed an association between traumatic brain injury (TBI) and subsequent glioblastoma (GBM) formation.
Methods:We used literature searches and radiographic evidence from two patients to assess the possibility of a link between TBI and GBM.
Results:Epidemiological studies are equivocal on a possible link between brain trauma and increased risk of malignant glioma formation. We present two case reports of patients with GBM arising at the site of prior brain injury.
Conclusion:The hypothesis that TBI may predispose to gliomagenesis is disputed by several large-scale epidemiological studies, but supported by some. Radiographic evidence from two cases presented here suggest that GBM formed at the site of brain injury. We propose a putative pathogenesis model that connects post-traumatic inflammation, stem and progenitor cell transformation, and gliomagenesis.
Keywords: Brain tumor, glioblastoma, traumatic brain injury
INTRODUCTION
Glioblastoma (GBM) is the most common and deadly brain malignancy, with over 10000 new cases in the US annually and a median survival of only 14–16 months after surgical resection and concurrent chemoradiotherapy.[
Environmental risk factors for GBM remain poorly defined, with the exception of exposure to ionizing radiation.[
The injury must be severe enough to cause a tissue repair process to commence; The area of the traumatic injury should correspond directly with the location of the subsequent GBM; There should be a gap of at least 1 year between the injury to the brain and the appearance of the tumor. A longer latent period is considered to be a stronger evidence of a causal relationship.
Here, we present two patients who developed GBM at the exact same site where they had suffered TBI several years ago. We also review relevant epidemiological studies, which are overall equivocal on the association between TBI and GBM. Finally, we propose a mechanism that could explain the biological link between TBI and gliomagenesis by invoking inflammation as the trigger for oncogenic transformation of neural stem and progenitor cells that migrate to the injured tissue for repair. Chronic inflammation has been shown to be a major predisposing factor for many solid tumors, including, but not limited to, hepatocellular carcinoma and colon cancer.[
MATERIALS AND METHODS
We performed literature searches on PubMed to identify epidemiological studies that investigated the link between brain injury and GBM formation. We also collected and analyzed clinical and radiographic data from two patients with a history of TBI, who subsequently developed GBM.
RESULTS
Epidemiological studies
Several epidemiological studies have attempted to analyze the association between GBM formation and the presence of previous TBI. These studies present varying results regarding the strength of correlation and odds ratio for TBI-induced GBM. Nygren et al. performed a population-based cohort study in Sweden, which analyzed over 300,000 patients who were documented to have suffered traumatic brain injury between 1965 and 1994.[
A Taiwanese group analyzed a cohort of 5007 patients who visited ambulatory care centers between 2001 and 2002.[
Gurney et al. performed a multicenter case-control study in which the association between brain injury and tumor formation was analyzed specifically in children.[
A study done by Preston-Martin et al.[
CASE REPORTS
Patient 1
This patient is a 65-year-old left-handed man, who suffered serious head trauma causing a large left frontal contusion at age 54 after a fall at the workplace. At that time, he was placed in a medically induced coma for 3 days. Serial imaging after the injury demonstrated encephalomalacia and gliosis at the site of the contusion [
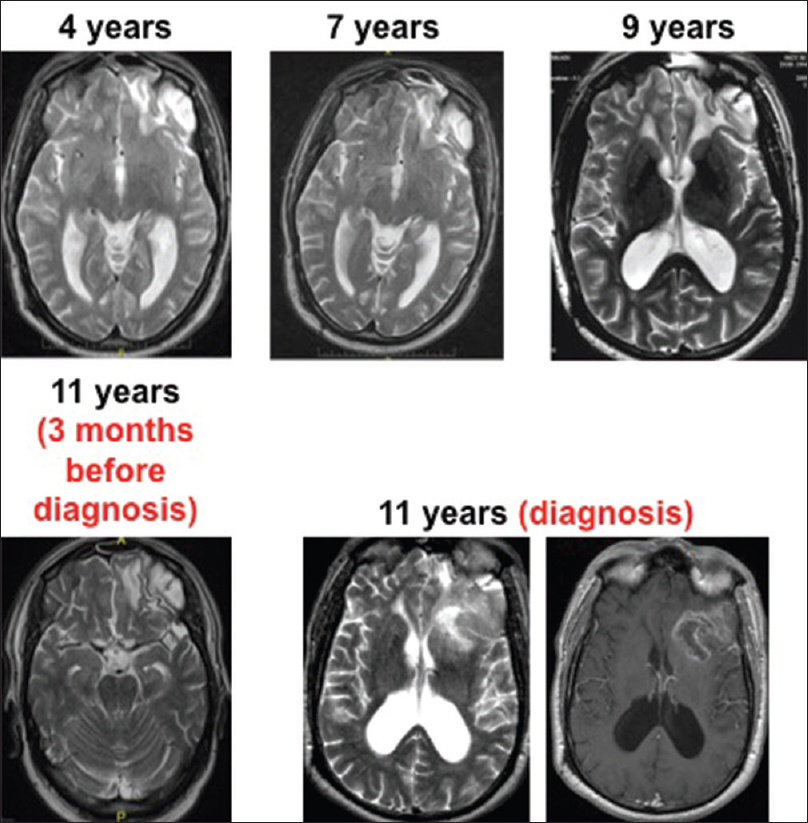
In preparation for resection of the tumor, a Wada Test revealed bilateral hemispheric language dominance. To avoid damaging language-processing centers, awake language mapping was attempted during planned operative resection. The patient became agitated during language mapping and the surgeon opted to place subdural grid electrodes for extraoperative speech mapping, which revealed no regions associated with language function in the vicinity of the tumor. In a second operative procedure under general anesthesia, he underwent uneventful resection of >90% of the tumor with residual in the left insula. Histopathologic analysis confirmed the diagnosis of GBM. The Ki-67 immunolabeling index using MIB1 antibody was up to 25%. Immunohistochemistry for the R132H mutation in IDH1 was negative.
After surgery, he was treated with concurrent chemoradiotherapy (Stupp protocol). However, the tumor recurred rapidly and he expired within 4 months after surgery.
Patient 2
This patient is a 54-year-old right-handed man who suffered a large contusion and severe injury to the inferior right frontal lobe in an automobile accident at age 47 [
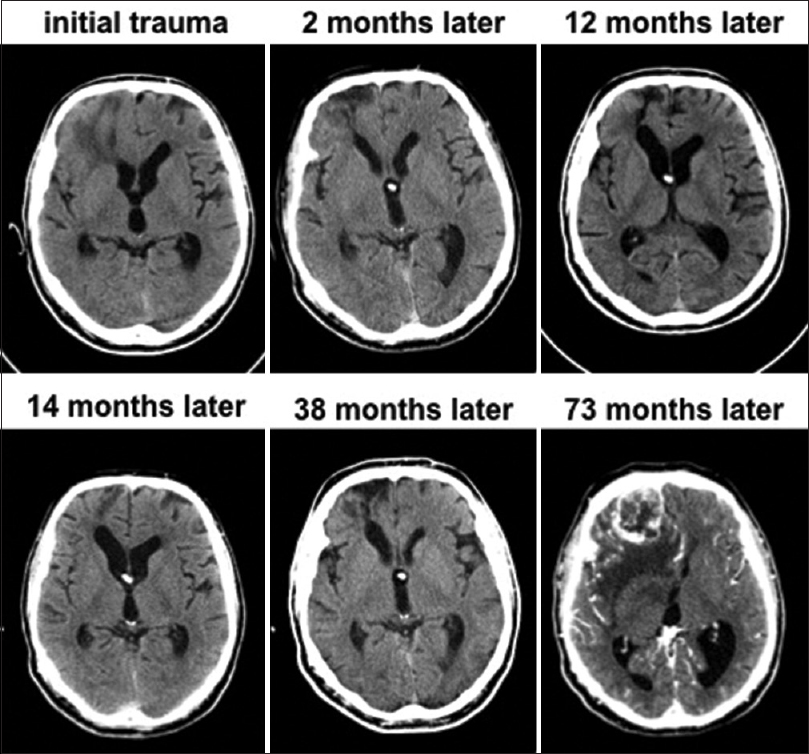
Seven years after the injury, he came again to medical attention due to headaches and confusion. CT imaging indicated a 4 cm heterogeneously enhancing inferior right frontal mass at the site of the brain injury. There was extensive vasogenic edema surrounding the lesion. He underwent right frontal craniotomy for gross total resection of the mass, which was diagnosed as GBM. Immunohistochemistry for the R132H mutation in IDH1 was negative. He was subsequently treated with chemoradiotherapy as per Stupp protocol. As per the most recent follow-up 8 months post-resection, he remains neurologically intact with no evidence for recurrence on CT.
DISCUSSION
Epidemiological studies are largely equivocal on the link between TBI and subsequent formation of malignant glioma. In fact, large-scale studies have shown no correlation. However, the two presented cases show GBM formation at the site of prior brain injury. Our hypothesis is that, in some patients, an underlying biological vulnerability predisposes them to gliomagenesis after brain trauma. What are the mechanisms that could potentially explain injury-driven gliomagenesis?
We propose that the inflammatory response that ensues after TBI is linked to oncogenic transformation of neural stem and progenitor cells that chemotactically migrate to the injured site in response to inflammation [
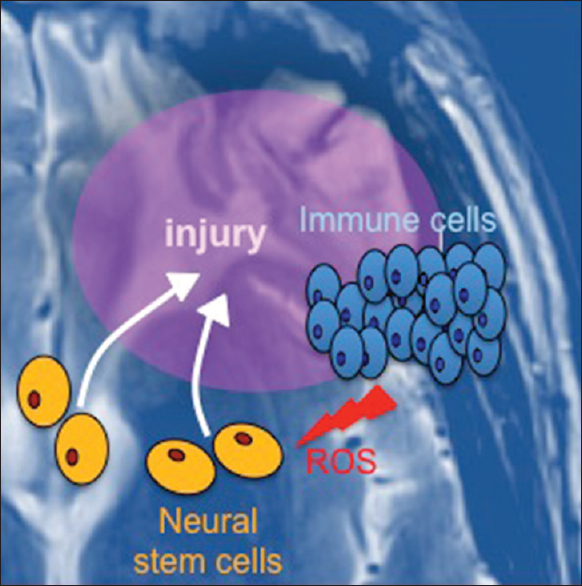
Figure 3
Model linking traumatic brain injury to glioblastoma formation. Upon injury to the brain, neural stem cells migrate to the site to enable tissue repair. At the same time, immune cells are recruited. Immune cells release reactive oxygen species (ROS), which can induce mutagenesis and initiate oncogenic transformation of stem cells
Inflammation
Inflammation at the site of brain injury is well-documented in the literature.[
A number of inflammatory cytokines, including IL-1, TNF, IL-10, IL-6, IL-8, and MCP-1, are known to be upregulated in the context of TBI and facilitate recruitment of myeloid immune cells.[
Stem cell migration to injury site
Neural stem cells in the adult brain localize primarily to two neurogenic areas, namely, the subventricular zone of the lateral ventricles and the subgranular zone of the hippocampal dentate gyrus (for review see[
The oncogenic potential of neural stem cells has been suggested by the observation that neurogenic niches in the brain are sensitive to chemical oncogenesis.[
CONCLUSION
In conclusion, large-scale epidemiological studies have not shown a definitive link between TBI and increased risk of developing GBM. However, the two patients presented here developed GBM at the site of their brain injury several years later. It is, therefore, possible that an underlying biological vulnerability in a subset of patients with TBI may predispose them to gliomagenesis. We propose a putative model that links neuroinflammation to mutagenesis in neural stem and progenitor cells migrating to the site of injury, leading to their neoplastic transformation and glioma initiation. In the future, as molecular mechanisms of gliomagenesis and the brain's response to TBI become clearer, we hope to identify the biological mechanisms that make a subset of patients susceptible to brain tumor formation after injury.
Financial support and sponsorship
N. Sumru Bayin received support from NYSTEM Institutional training grant #CO26880. Dimitris G. Placantonakis received support from NIH/NINDS 1R21NS087241-01, NIH/NINDS 1R21NS088775-01, NIH/NINDS 1R03NS087349-01, NIH/NCI 2P30CA016087-33, NIH/NCATS UL1 TR000038, NYU Cancer Institute, NYU Clinical and Translational Science Institute and the B*Cured Foundation.
Conflicts of interest
There are no conflicts of interest.
References
1. Adeberg S, König L, Bostel T, Harrabi S, Welzel T, Debus J. Glioblastoma recurrence patterns after radiation therapy with regard to the subventricular zone. Int J Radiat Oncol Biol Phys. 2014. 90: 886-93
2. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nature Genet. 2011. 43: 1262-5
3. Anselmi E, Vallisa D, Bertè R, Vanzo C, Cavanna L. Post-traumatic glioma: Report of two cases. Tumori. 2006. 92: 175-7
4. Barnabé-Heider F, Göritz C, Sabelström H, Takebayashi H, Pfrieger FW, Meletis K. Origin of new glial cells in intact and injured adult spinal cord. Cell Stem Cell. 2010. 7: 470-82
5. Bohman LE, Swanson KR, Moore JL, Rockne R, Mandigo C, Hankinson T. Magnetic resonance imaging characteristics of glioblastoma multiforme: Implications for understanding glioma ontogeny. Neurosurgery. 2010. 67: 1319-27
6. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR. The somatic genomic landscape of glioblastoma. Cell. 2013. 155: 462-77
7. Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008. 105: 3581-6
8. . Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008. 455: 1061-8
9. Carlén M, Meletis K, Göritz C, Darsalia V, Evergren E, Tanigaki K. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci. 2009. 12: 259-67
10. Chaichana KL, McGirt MJ, Frazier J, Attenello F, Guerrero-Cazares H, Quinones-Hinojosa A. Relationship of glioblastoma multiforme to the lateral ventricles predicts survival following tumor resection. J Neurooncol. 2008. 89: 219-24
11. Chen YH, Keller JJ, Kang JH, Lin HC. Association between traumatic brain injury and the subsequent risk of brain cancer. J Neurotrauma. 2012. 29: 1328-33
12. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002. 420: 860-7
13. Curran CS, Bertics PJ. Eosinophils in glioblastoma biology. J Neuroinflammation. 2012. 9: 11-
14. Di Trapani G, Carnevale A, Scerrati M, Colosimo C, Vaccario ML, Mei D. Post-traumatic malignant glioma. Report of a case. Ital J Neurol Sci. 1996. 17: 283-6
15. Elvira G, García I, Gallo J, Benito M, Montesinos P, Holgado-Martin E. Detection of mouse endogenous type B astrocytes migrating towards brain lesions. Stem Cell Res. 2015. 14: 114-29
16. Fredens K, Dahl R, Venge P. The Gordon phenomenon induced by the eosinophil cationic protein and eosinophil protein X. J Allergy Clin Immunol. 1982. 70: 361-6
17. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012. 338: 1080-4
18. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014. 370: 699-708
19. Goings GE, Sahni V, Szele FG. Migration patterns of subventricular zone cells in adult mice change after cerebral cortex injury. Brain Res. 2004. 996: 213-26
20. Gurney JG, Preston-Martin S, McDaniel AM, Mueller BA, Holly EA. Head injury as a risk factor for brain tumors in children: Results from a multicenter case-control study. Epidemiology. 1996. 7: 485-9
21. Halliday JJ, Holland EC. Connective tissue growth factor and the parallels between brain injury and brain tumors. J Natl Cancer Inst. 2011. 103: 1141-3
22. Hopewell JW. The subependymal plate and the genesis of gliomas. J Pathol. 1975. 117: 101-3
23. Inskip PD, Mellemkjaer L, Gridley G, Olsen JH. Incidence of intracranial tumors following hospitalization for head injuries (Denmark). Cancer Causes Control. 1998. 9: 109-16
24. Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003. 24: 171-89
25. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013. 136: 28-42
26. Koos WT, Horaczek A. Statistics of intracranial midline tumors in children. Acta Neurochir Suppl. 1985. 35: 1-5
27. Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008. 8: 384-92
28. Lang B, Liu HL, Liu R, Feng GD, Jiao XY, Ju G. Astrocytes in injured adult rat spinal cord may acquire the potential of neural stem cells. Neuroscience. 2004. 128: 775-83
29. Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013. 8: e68963-
30. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011. 146: 209-21
31. Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009. 15: 110-6
32. Mc DA, Opeskin K. Timing of early changes in brain trauma. Am J Forensic Med Pathol. 1998. 19: 1-9
33. Modrek AS, Bayin NS, Placantonakis DG. Brain stem cells as the cell of origin in glioma. World J Stem Cells. 2014. 6: 43-52
34. Monteiro GT, Pereira RA, Koifman RJ, Koifman S. Head injury and brain tumours in adults: A case-control study in Rio de Janeiro, Brazil. Eur J Cancer. 2006. 42: 917-21
35. Mossman BT. Introduction to serial reviews on the role of reactive oxygen and nitrogen species (ROS/RNS) in lung injury and diseases. Free Radic Biol Med. 2003. 34: 1115-6
36. Munch TN, Gørtz S, Wohlfahrt J, Melbye M. The long-term risk of malignant astrocytic tumors after structural brain injury--A nationwide cohort study. Neuro Oncol. 2015. 17: 718-24
37. Murray KN, Parry-Jones AR, Allan SM. Interleukin-1 and acute brain injury. Front Cell Neurosci. 2015. 9: 18-
38. Nygren C, Adami J, Ye W, Bellocco R, af Geijerstam JL, Borg J. Primary brain tumors following traumatic brain injury--A population-based cohort study in Sweden. Cancer Causes Control. 2001. 12: 733-7
39. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: A clinical review. JAMA. 2013. 310: 1842-50
40. Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nature Genet. 2011. 43: 1256-61
41. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006. 9: 157-73
42. Preston-Martin S, Yu MC, Henderson BE, Roberts C. Risk factors for meningiomas in men in Los Angeles County. J Natl Cancer Inst. 1983. 70: 863-6
43. Rajaraman P, Melin BS, Wang Z, McKean-Cowdin R, Michaud DS, Wang SS. Genome-wide association study of glioma and meta-analysis. Hum Genet. 2012. 131: 1877-88
44. Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005. 353: 811-22
45. Schwartzbaum J, Ahlbom A, Malmer B, Lönn S, Brookes AJ, Doss H. Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res. 2005. 65: 6459-65
46. Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009. 41: 899-904
47. Simon M, Hosking FJ, Marie Y, Gousias K, Boisselier B, Carpentier C. Genetic risk profiles identify different molecular etiologies for glioma. Clin Cancer Res. 2010. 16: 5252-9
48. Tong J, Latzman JM, Rauch J, Zagzag DS, Huang JH, Samadani U. Sonic hedgehog agonist fails to induce neural stem cell precursors in a porcine model of experimental intracranial hemorrhage. Acta Neurochir Suppl. 2011. 111: 151-4
49. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010. 17: 98-110
50. Vick NA, Lin MJ, Bigner DD. The role of the subependymal plate in glial tumorigenesis. Acta Neuropathol. 1977. 40: 63-71
51. Wibom C, Ghasimi S, Van Loo P, Brännström T, Trygg J, Lau C. EGFR gene variants are associated with specific somatic aberrations in glioma. PLoS One. 2012. 7: e47929-
52. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013. 4: 18-
53. Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nature Genet. 2009. 41: 905-8
54. Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci. 2006. 26: 6627-36
55. Yang TH, Kon M, Hung JH, Delisi C. Combinations of newly confirmed Glioma-Associated loci link regions on chromosomes 1 and 9 to increased disease risk. BMC Med Genomics. 2011. 4: 63-
56. Zhou B, Liu W. Post-traumatic glioma: Report of one case and review of the literature. Int J Med Sci. 2010. 7: 248-50

