

- Department of Neurosurgery, Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan
- Department of Neurosurgery, Toyama City Hospital, Toyama, Japan
Correspondence Address:
Yasuhiko Hayashi
Department of Neurosurgery, Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan
DOI:10.4103/2152-7806.185773
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Oishi M, Hayashi Y, Fukui I, Kita D, Miyamori T, Nakada M. Xanthomatous hypophysitis associated with autoimmune disease in an elderly patient: A rare case report. Surg Neurol Int 07-Jul-2016;7:
How to cite this URL: Oishi M, Hayashi Y, Fukui I, Kita D, Miyamori T, Nakada M. Xanthomatous hypophysitis associated with autoimmune disease in an elderly patient: A rare case report. Surg Neurol Int 07-Jul-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/xanthomatous-hypophysitis-associated-autoimmune-disease-elderly-patient-rare-case-report/
Abstract
Background:Xanthomatous hypophysitis (XH) is an extremely rare form of primary hypophysitis characterized by infiltration of the pituitary gland by mixed types of inflammatory cells, including foamy cells, plasma cells, and small mature lymphocytes. XH manifests as varying degrees of hypopituitarism. Although several previous reports have denied a possible contribution of autoimmune mechanism, the exact pathogenesis of XH remains unclear.
Case Description:We describe the case of a 72-year-old woman with a history of rheumatoid arthritis and Sjögren's syndrome who presented with panhypopituitarism and diabetes insipidus. At the time of her visit, she also experienced relapsed rheumatoid arthritis and Sjögren's syndrome, manifesting as arthralgia. Magnetic resonance imaging (MRI) showed a multicystic mass in the sellar and suprasellar regions. In the course of steroid replacement therapy for hypocortisolism, the patient's arthralgia diminished, and MRI revealed shrinkage of the mass. XH was diagnosed histologically following a transsphenoidal endoscopic biopsy, and it was the oldest case of XH.
Conclusion:To the best of our knowledge, this patient is the oldest of reported patients diagnosed with XH. Steroid therapy may be effective to XH temporarily. XH should be considered when diagnosing pituitary cystic lesions in elderly patients with autoimmune disease.
Keywords: Autoimmune disease, hypopituitarism, xanthomatous hypophysitis
INTRODUCTION
Primary hypophysitis is an inflammation of the pituitary gland that is histologically characterized by the focal or diffuse infiltration of inflammatory cells and destruction of the pituitary gland, resulting in varying degrees of hypopituitarism. Primary hypophysitis has been classified into three distinct histological subtypes: Lymphocytic, granulomatous, and xanthomatous hypophysitis (XH).[
We describe the case of an elderly woman with a history of rheumatoid arthritis and Sjögren's syndrome who presented with hypopituitarism and diabetes insipidus (DI) and was histologically diagnosed with XH.
CASE REPORT
A 72-year-old woman had experienced polydipsia, polyuria, fatigue, low-grade fever, and arthralgia for several months before visiting our department. Her medical history included rheumatoid arthritis and Sjögren's syndrome. Six years prior to her visit, she was treated for 6 months with 5 mg of prednisolone per day, and her arthralgia diminished. Her family history was unremarkable. Results of a physical examination were normal except for the presence of arthralgia. Endocrine laboratory testing revealed a moderately elevated serum prolactin (PRL) level of 95.2 ng/mL (normal range: 6.1–30.5 ng/mL), suggesting a compression of the pituitary stalk. Test results of other hormones secreted from the anterior lobe of the pituitary gland were as follows: Thyroid-stimulating hormone (TSH): 0.54 μU/mL (normal range: 0.35–4.94 μU/mL); free T4: 1.25 ng/dL (normal range: 0.70–1.48 ng/dL); adrenocorticotrophic hormone (ACTH): 15.2 pg/mL (normal range: 7.2–63.3 pg/mL); cortisol: 3.6 μg/dL (normal range: 3.8–18.4 μg/dL); growth hormone (GH): 0.30 ng/mL (normal range: 0.28–1.64 ng/mL); luteinizing hormone: 0.42 mIU/mL; and follicle stimulating hormone: 3.28 mIU/mL. Thyrotropin-releasing hormone stimulation resulted in an appropriate increase in PRL level to 132.6 ng/mL after 15 min and in TSH level to 10.11 μU/mL as peak values. An insulin-induced hypoglycemia test resulted in poor responses of both GH and cortisol levels (peak values: 0.79 ng/mL and 3.2 μg/dL, respectively). These results indicated hypopituitarism affecting ACTH, gonadotropin, and GH axes. Based on the results of hypertonic saline infusion test, the patient was diagnosed with the central type of DI. Hypocortisolism was treated with 20 mg of hydrocortisone for 2 months. The dose was then gradually decreased to 5 mg. About 0.5 μg of 1-deamino-8-D-arginine vasopressin (DDAVP) per day was taken for DI as hormone replacement therapy. The patient showed prompt recovery from her symptoms, including arthralgia.
Magnetic resonance imaging (MRI) showed a thickened pituitary stalk with a multicystic lesion extending toward the hypothalamus and no evidence of posterior lobe hyperintensity on T1-weighted imaging [Figure
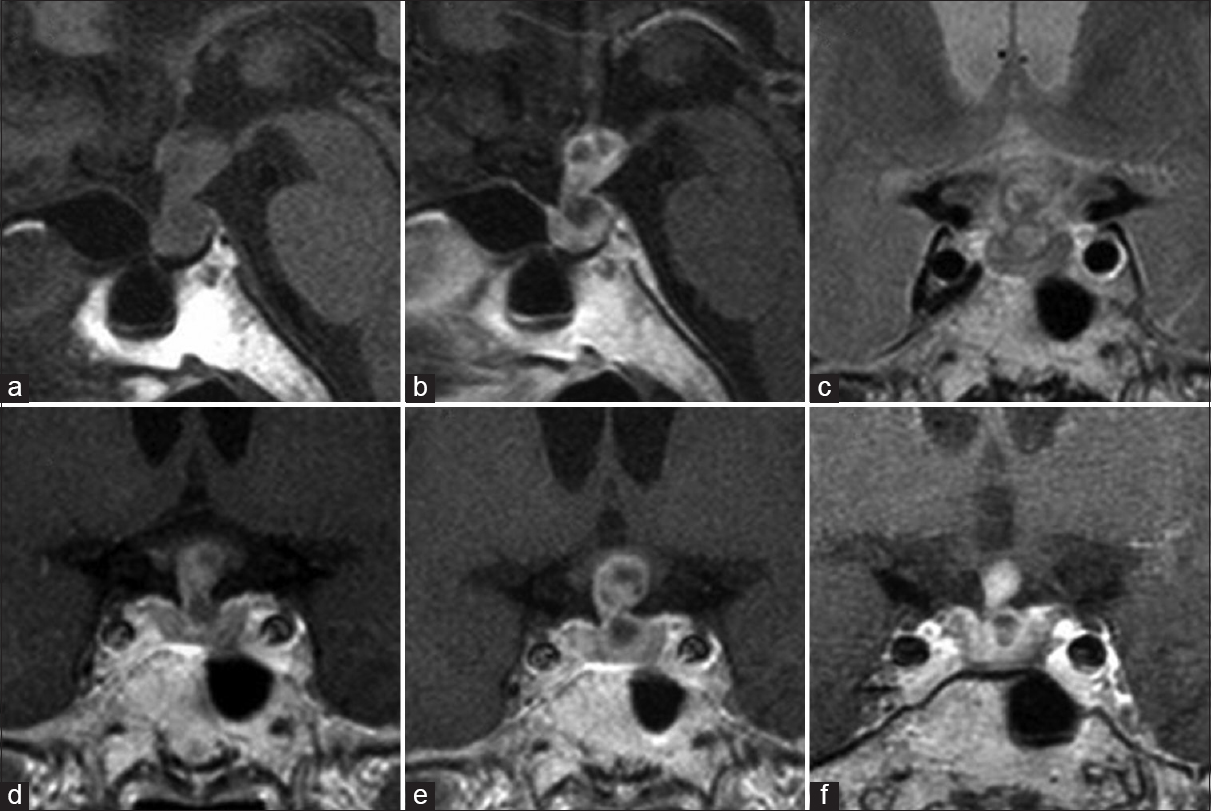
Figure 1
Magnetic resonance imaging from onset to surgery. Sagittal section of T1-weighted images on admission showed thickened pituitary and stalk extending toward the hypothalamus and no evidence of posterior lobe hyperintensity (a). Sagittal and coronal section of gadolinium-enhanced T1-weighted image (b and e) and coronal section of T2-weighted image (c) at the time of her visit showed the intrasellar and suprasellar multicystic lesion with heterogeneous contrast. Coronal section of gadolinium-enhanced T1-weighted image showed the lesion shrinking (f) after transient expanding on admission (e) compared to 1 month ago (d)
Endoscopic exploration clearly displayed a soft yellowish lesion along with the pituitary stalk [
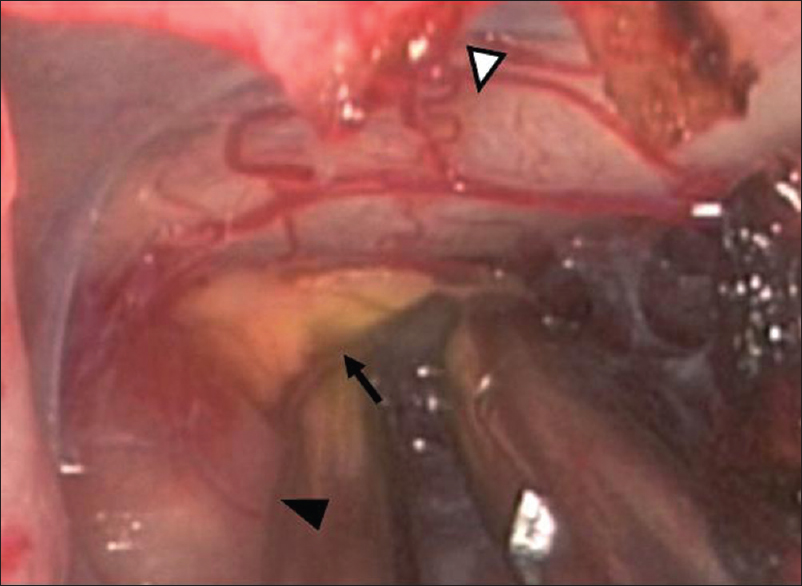
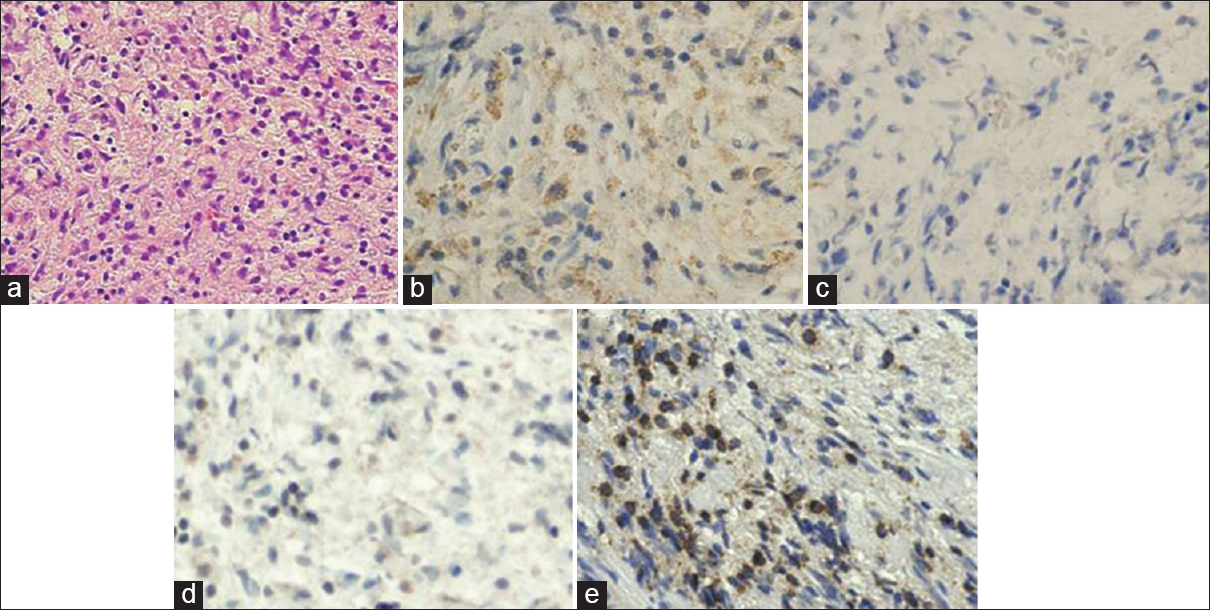
Figure 3
Pathological findings of the lesion. The cystic lesion was histologically characterized by infiltration of the anterior pituitary by foamy histiocytes, plasma cells, and lymphocytes (a, ×200). The foamy cells were immunopositive for the macrophage marker CD68 (b, ×200) and immunonegative for CD1a (c, ×200) and S-100 protein (d, ×200). The majority of lymphocytes are immunopositive for CD3 (e, ×200). There was no evidence of pituitary adenoma or granuloma or necrosis
The patient was discharged without any neurological complications. Postoperative pituitary function was unchanged compared to the preoperative state. The patient continued to take 5 mg of hydrocortisone and 0.5 μg of DDAVP per day. Postoperative MRI showed no progression of the lesion [
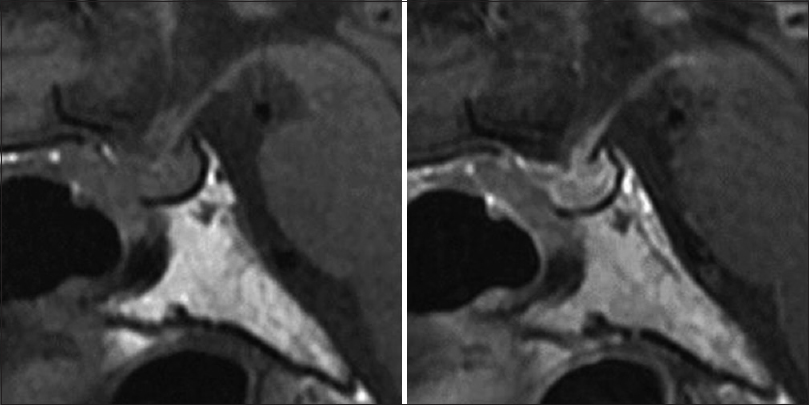
DISCUSSION
XH is the rarest form of primary hypophysitis and is histologically characterized by the diffuse infiltration of the pituitary gland by mixed inflammatory cells, including foamy cells, plasma cells, small mature lymphocytes, and multi-nucleated giant cells.[
Transsphenoidal surgery is a useful diagnostic and therapeutic tool for the clinical entities discussed here and should be performed in patients whose lesions show progression of clinical or neuroradiological findings. Indeed, surgical intervention and histological verification have been performed in all of the 19 previously reported cases of XH.[
Steroid therapy has been reported to temporarily relieve inflammation in some patients with primary hypophysitis, particularly lymphocytic hypophysitis, which is believed to arise from an autoimmune mechanism. Gutenberg et al. reported that one patient with XH who was treated with methylprednisolone did not show any improvement.[
Aste et al. reported a case of XH with ulcerative colitis[
Our patient had a history of rheumatoid arthritis and Sjögren's syndrome and had received steroid therapy for 6 months. When she presented with initial endocrinological symptoms indicative of XH, steroid therapy was suspended and arthralgia attributed to rheumatoid arthritis returned. However, after initiating hormone replacement therapy, the patient's symptoms rapidly improved, and MRI revealed gradual shrinkage of the suprasellar lesion during the 2 months after onset. Although there are limited data to support an autoimmune etiology, we speculate that autoimmune disease may not only be associated with the development of XH, but may also reflect its activity.
XH has been shown to be highly prevalent in young women. The mean age of clinical onset among previously reported cases is 37.4 ± 14.8 years. Our patient, a 72-year-old female, is the oldest of all previously reported cases. If an autoimmune mechanism contributes to the development of XH, as described above, the age of XH onset may correspond to that of the associated autoimmune disease. In support of this hypothesis, the XH patients described above with ulcerative colitis and Hashimoto's thyroiditis were 31- and 36-year-old, respectively, near the peak ages of onset for ulcerative colitis (20–30 years) and Hashimoto's thyroiditis (35–55 years).[
CONCLUSION
XH occurs less frequently than the other two types of primary hypophysitis. Similar to lymphocytic hypophysitis, XH may be associated with autoimmune diseases. If cystic lesions along the pituitary stalk are detected in elderly patients with hypopituitarism and autoimmune disease such as rheumatoid arthritis, XH should be considered as a possible diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Aste L, Bellinzona M, Meleddu V, Farci G, Manieli C, Godano U. Xanthomatous hypophysitis mimicking a pituitary adenoma: Case report and review of the literature. J Oncol. 2010. p.
2. Burt MG, Morey AL, Turner JJ, Pell M, Sheehy JP, Ho KK. Xanthomatous pituitary lesions: A report of two cases and review of the literature. Pituitary. 2003. 6: 161-8
3. Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab. 2001. 86: 1048-53
4. Deodhare SS, Bilbao JM, Kovacs K, Horvath E, Nomikos P, Buchfelder M. Xanthomatous hypophysitis: A novel entity of obscure etiology. Endocr Pathol. 1999. 10: 237-41
5. Folkerth RD, Price DL, Schwartz M, Black PM, De Girolami U. Xanthomatous hypophysitis. Am J Surg Pathol. 1998. 22: 736-41
6. Gutenberg A, Buslei R, Fahlbusch R, Buchfelder M, Brück W. Immunopathology of primary hypophysitis: Implications for pathogenesis. Am J Surg Pathol. 2005. 29: 329-38
7. Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Brück W, Caturegli P. Primary hypophysitis: Clinical-pathological correlations. Eur J Endocrinol. 2006. 155: 101-7
8. Haas AV, Smith TW. Xanthomatous hypophysitis: An unusual case of a sellar mass. Neurol Bull. 2014. 4: 41-4
9. Hanna B, Li YM, Beutler T, Goyal P, Hall WA. Xanthomatous hypophysitis. J Clin Neurosci. 2015. 22: 1091-7
10. Joung JY, Jeong H, Cho YY, Huh K, Suh YL, Kim KW. Steroid responsive xanthomatous hypophysitis associated with autoimmune thyroiditis: A case report. Endocrinol Metab (Seoul). 2013. 28: 65-9
11. Niyazoglu M, Celik O, Bakkaloglu DV, Oz B, Tanriöver N, Gazioglu N. Xanthomatous hypophysitis. J Clin Neurosci. 2012. 19: 1742-4
12. Tang F, Liu H, Zhou S, Liu J, Xiao E, Tan C. MRI manifestation of xanthomatous hypophysitis: A case report and review of the literature. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2015. 40: 228-32
13. Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H. Spectrum of different types of hypophysitis: A clinicopathologic study of hypophysitis in 31 cases. Endocr Pathol. 2002. 13: 183-95

