

- Departments of Neurosurgery, University Hospital Dubrava, Zagreb, Croatia.
- Departments of Pathology and Cytology, University Hospital Dubrava, Zagreb, Croatia.
Correspondence Address:
Marina Raguz
Departments of Neurosurgery, University Hospital Dubrava, Zagreb, Croatia.
DOI:10.25259/SNI_288_2019
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Fadi Almahariq, Marina Raguz, Dominik Romic, Domagoj Dlaka, Darko Oreskovic, Patricija Sesar, Darko Chudy. A biphasic tumor in posterior cranial fossa and the pineal region in young adult. 11-Apr-2020;11:64
How to cite this URL: Fadi Almahariq, Marina Raguz, Dominik Romic, Domagoj Dlaka, Darko Oreskovic, Patricija Sesar, Darko Chudy. A biphasic tumor in posterior cranial fossa and the pineal region in young adult. 11-Apr-2020;11:64. Available from: https://surgicalneurologyint.com/surgicalint-articles/9957/
Date of Submission
06-May-2019
Date of Acceptance
30-Mar-2020
Date of Web Publication
11-Apr-2020
Abstract
Background: Biphasic tumors of the central nervous system are rarely described and mainly consisted out of the glial and mesenchymal component. The tumor originated out of both astrocytes and pinealocytes, best to our knowledge, has not been described. We present a case of a brain tumor consisted out of pilocytic astrocytoma (PA) and pineocytoma as components situated in the pineal region and posterior cranial fossa in young adult.
Case Description: We present a 21-year-old patient with a history of intermittent headache, followed by nausea and vomiting, double vision, and dextropulsion. Magnetic resonance imaging revealed an extensive cystic-solid expansive formation in the posterior cranial fossa with a solid part in the area of the pineal gland. The patient underwent surgical resection. The pathohistological analysis showed two types of tumor cells; the major part of tumor showed features of PA, while minor part corresponded to pineocytoma.
Conclusion: PA accounts for 5% of all gliomas and is most common in children and young adults. It usually occurs in the cerebellum, the optic pathway, third ventricular region, etc. Pineocytomas are rare, accounting up to 1% of all intracranial tumors. Since tumors origin is different, there must be complex molecular events or mutations that can lead to cell rearrangements and generation of two histologically different tissues in the same tumor mass. The course of treatment options is different for PA and pineocytoma; therefore, the case of brain mass consisted out of two different tissues can be helpful when deciding about the treatment of tumors in posterior cranial fossa and pineal region.
Keywords: Biphasic tumor, Brain tumor, Pilocytic astrocytoma, Posterior cranial fossa, Pyneocitoma
INTRODUCTION
The central nervous system (CNS) biphasic tumors occur rarely. According to the literature, only few cases of biphasic tumor have been described.[
CASE DESCRIPTION
History
A 21-year-old male patient presented with a 2 weeks history of intermittent headache followed by nausea and vomiting, double vision, discrete horizontal nystagmus bilaterally, and discrete dextropulsion. Initial computerized tomography (CT) and magnetic resonance imaging (MRI) revealed an extensive cystic-solid expansive formation in the posterior cranial fossa with a solid part in the area of the pineal gland. After the administration of intravenous contrast, the solid mass showed heterogeneous enhancement [
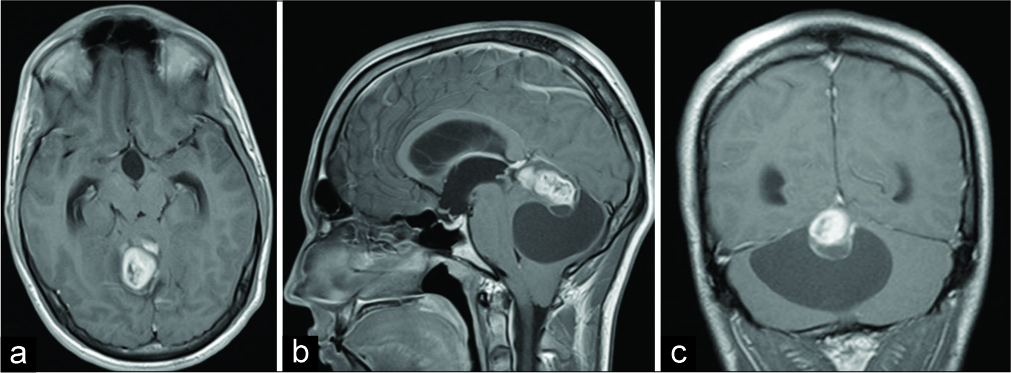
Figure 1:
Initial brain magnetic resonance imaging showing right- sided extra-axial high parietal parasagittal mass in (a) transverse plain, T1-weighted image; (b) sagittal plain, T1-weighted image; and (c) coronal plain, T1-weighted image. Note the marginal heterogeneous enhancement of the tumor after the administration of intravenous contrast.
Operation
The patient underwent a paramedian occipital and suboccipital craniotomy and total tumor removal. The surgery went without any complications. A control CT scan performed on the 1st postoperative day, revealed a satisfactory finding without any signs of ischemia, hemorrhage, or residual tumor. The patient recovered uneventfully. Tissue samples acquired during the surgical procedure underwent pathohistological and cytological analysis. Cytological analysis showed no signs of malignant cells. The pathohistological analysis showed two types of tumor cells. A major part of the tumor, more than 75% of total tumor tissue, consisted out of compacted bipolar piloid astrocyte. Focal microcystic changes were observed. The vascular network was pronounced, and the mitosis was not found. Scattered eosinophilic granular bodies and Rosenthal fibers were observed under higher magnification. Proliferation index Ki67 was up to 3% diffusely and up to 10% focally [
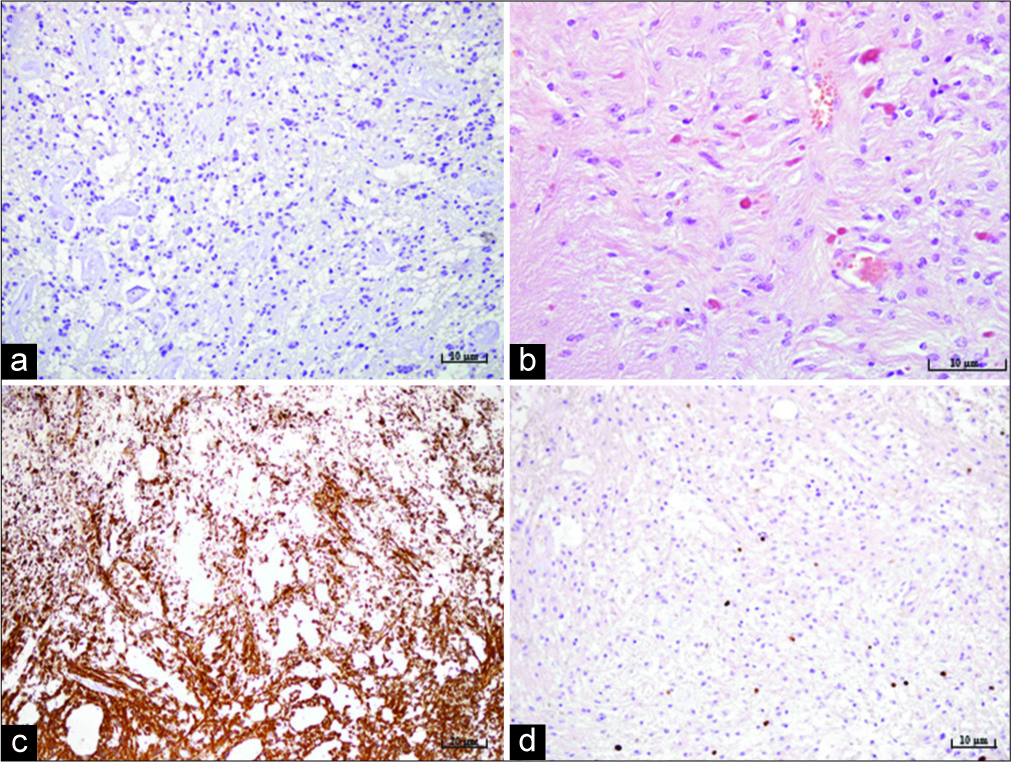
Figure 2:
Microphotography of a pathohistological section showing histological and immunohistochemical features of a pilocytic astrocytoma stained with (a) IDH-1, original magnification of ×200, showing the absence of IDH mutation, (b) hematoxylin and eosin, original magnification of ×400, showing characteristic Rosenthal fibers and eosinophilic granular bodies, (c) strong positive reaction for glial fibrillary acidic protein staining, original magnification of ×100, (d) proliferation index Ki67, original magnification of ×200, showing up to 3% diffusely and up to 10% focally.
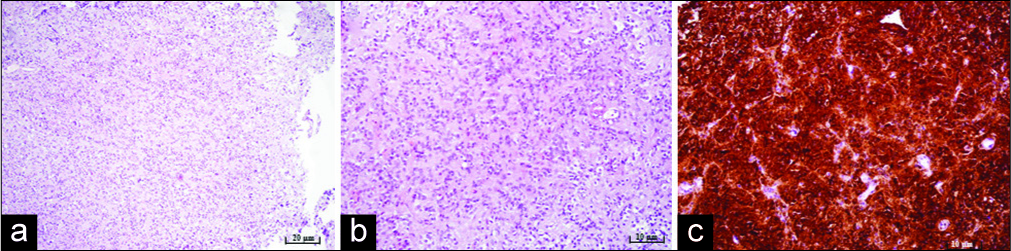
Figure 3:
Microphotography of a pathohistological section showing histological and immunohistochemical features of a pineocytoma stained with (a) hematoxylin and eosin, original magnification of ×100, showing moderate cellularity, (b) hematoxylin and eosin, original magnification of ×200, showing pineocytomas rosette formation, (c) strong positive reaction for synaptophysin immunohistochemistry staining, original magnification of ×200.
Postoperative course
Five months afterward, an MRI and neurosurgical control was performed. The patient was without any neurological deficits. MRI revealed a satisfying postoperative result without the recurrence of intracranial masses [
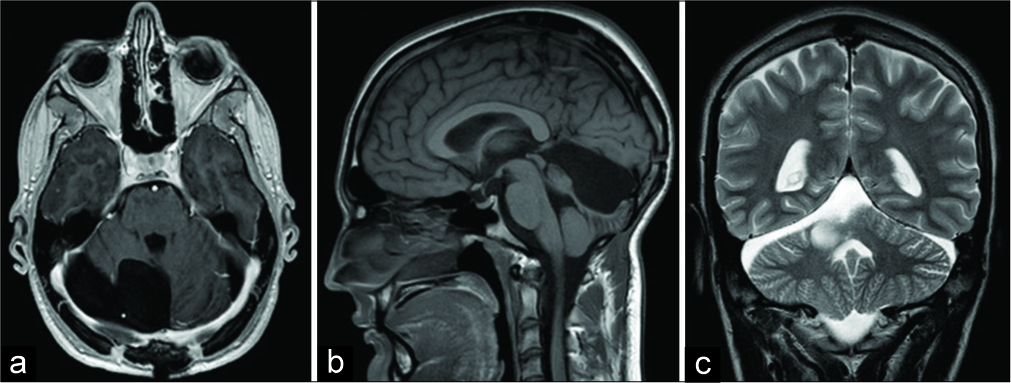
DISCUSSION
Biphasic tumors of the CNS are rarely described and mainly consisted out of the glial and mesenchymal component.[
PA, clinically referred to by several terms, including cerebellar astrocytoma, optic glioma, and infundibuloma, is a rare glioma, classified as Grade I by the WHO. PA accounts for 15.4% of children and adolescents and 17.6% of childhood primary brain tumors. Nevertheless, PA may occur at any age, becoming increasingly uncommon with advancing years. PA most frequently occurs in the cerebellum, followed by the supratentorial compartment, the optic pathway, third ventricular/hypothalamic region, brainstem, and the spinal cord.[
Gross examination of PA shows relatively soft-tissue tumor, white-grey in sections, as in major part of removed tumor. A cyst is usually present either within or around the tumor tissue, which can later result in a typical finding of cyst consisting of a tumor nodule. Patohistologically, PA is generally low to the moderate cellular tumor, characterized by a biphasic pattern with alternating densely and loosely packed areas. Micro cyst, granular bodies, and protoplasmic astrocyte-like cells (multipolar cells) with a round to oval nuclei can be in loose areas, while compact areas consist out of bipolar cells with the presence of Rosenthal fibers. Rosenthal fibers consist out of cells with long bipolar processes and elongated cytologically bland nuclei. Other characteristic includes vascular proliferation, sclerotic vessels, calcification, and chronic inflammation, while nuclear pleomorphism, mitosis, and necrosis are rare.[
As mentioned above, the major part of the removed tumor in our patient showed the pathohistological characteristic of PA. Patohistologically, pineocytomas are composed of small cells similar in appearance to normal pinealocytes, arranged in sheets, pineocytoma pseudo rosettes which are not characteristic and in normal pineal gland tissue. Immunohistochemically, they show a strong positive reaction to synaptophysin, neurofilament and neuron- specific enolase, while NeuN usually shows a negative reaction. Other neuronal markers, such as chromogranin A, are variable.[
PA is primarily treated by surgery, and the survival rates for patients are excellent. Radiotherapy or chemotherapy can be used as therapy of choice. In the case of pineocytomas, gross total resection is the appropriate treatment when anatomically possible because it is affiliated with long term survival. Adjuvant radiotherapy is a therapy of choice.
CONCLUSION
Since the course of treatment options are slightly different for PA and pineocytoma, we believe that this unique case of brain mass consisted out of two pathohistologically different tumors can be helpful when deciding about the treatment of tumors in posterior cranial fossa and pineal region.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Clark AJ, Sughrue ME, Aranda D, Parsa AT. Contemporary management of pineocytoma. Neurosurg Clin N Am. 2011. 22: 403-7
2. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015. 129: 775-88
3. Cyrine S, Sonia Z, Mounir T, Badderedine S, Kalthoum T, Hedi K. Pilocytic astrocytoma: A retrospective study of 32 cases. Clin Neurol Neurosurg. 2013. 115: 1220-5
4. Dutta G, Gupta R, Garg M, Singh D, Singh H, Srivastava AK. Giant parieto-occipital lobe pediatric gliosarcoma: Report of a rare entity and review of literature. Surg Neurol Int. 2018. 9: 111-
5. Fakhran S, Escott EJ. Pineocytoma mimicking a pineal cyst on imaging: True diagnostic dilemma or a case of incomplete imaging?. AJNR Am J Neuroradiol. 2008. 29: 159-63
6. Feigin IH, Gross SW. Sarcoma arising in glioblastoma of the brain. Am J Pathol. 1955. 31: 633-53
7. Jang SJ, Kim J, Cho JM, Noh S, Park SH, Kim SH. A biphasic tumor consisting of pilocytic astrocytoma with an anaplastic solitary fibrous tumor component in the pineal region: A case report and literature review. Neuropathology. 2013. 33: 288-91
8. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
9. Wade A, Hayhurst C, Amato-Watkins A, Lammie A, Leach P. Cerebellar pilocytic astrocytoma in adults: A management paradigm for a rare tumour. Acta Neurochir (Wien). 2013. 155: 1431-5


