

- Neurosurgery-Neurotraumatology Unit of University Hospital of Parma, Parma and Department of Neurosurgery, Reggio Emilia, Italy
- Department of Otolaryngology Unit of Institute for Scientific and Care Research “ASMN” of Reggio Emilia, Reggio Emilia, Italy
Correspondence Address:
D. Nasi
Neurosurgery-Neurotraumatology Unit of University Hospital of Parma, Parma and Department of Neurosurgery, Reggio Emilia, Italy
DOI:10.4103/sni.sni_450_16
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: D. Nasi, D. Perano, R. Ghadirpour, C. Iaccarino, F. Servadei, A. Romano. Primary pituitary neuroendocrine tumor: Case report and literature review. 05-Jun-2017;8:101
How to cite this URL: D. Nasi, D. Perano, R. Ghadirpour, C. Iaccarino, F. Servadei, A. Romano. Primary pituitary neuroendocrine tumor: Case report and literature review. 05-Jun-2017;8:101. Available from: http://surgicalneurologyint.com/surgicalint-articles/primary-pituitary-neuroendocrine-tumor-case-report-and-literature-review/
Date of Submission
13-Nov-2016
Date of Acceptance
22-Mar-2017
Date of Web Publication
05-Jun-2017
Abstract
Background:Neuroendocrine tumors (NET) originate from the diffuse neuroendocrine system. These can arise in almost every organ of the body, although they are most commonly found in the gastrointestinal tract and respiratory system. The skull base and sellar region are extremely rare sites for neuroendocrine carcinoma. Consequently, in this case, both diagnosis and definition of surgical goals, as well as further treatment strategies were challenging.
Case Description:A 65-year-old woman was admitted to our Neurosurgery Department with a rapidly progressive visus reduction, drowsiness, polyuria, and polydipsia. Neuroimaging showed a sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal compressing the optic chiasm and extending laterally toward the cavernous sinus. Differential diagnosis based on imaging included pituitary macroadenoma or metastasis. The patient underwent endoscopic endonasal transsphenoidal surgery. A total resection of the mass was impossible because of the infiltration of the optic chiasm and the intraoperative histological diagnosis of malignant epithelial neoplasm. Further histological evaluation revealed that the lesion was a NET with no other primary or metastatic sites detectable. Subsequently, the patient was successfully treated with fractioned stereotactic radiotherapy and polychemotherapy. Four years after the surgery, follow-up magnetic resonance imaging showed stability of the residual disease. Neurologic examination revealed a complete visual recovery.
Conclusions:Primary pituitary NET, though rare, should be included in the differential diagnosis of sellar lesions. A multimodality treatment approach is needed. Finally, the present case highlights, that in the case of a pituitary lesion infiltrating the optic chiasm, including NET, the endoscopic endonasal transsphenoidal subtotal resection followed by fractioned stereotactic radiotherapy and chemotherapy may represent an effective and safe choice of treatment.
Keywords: Endoscopic endonasal transsphenoidal approach, neuroendocrine tumor, pituitary, sellar region tumors
INTRODUCTION
This case report describes a neuroendocrine tumor (NET) arising from enterochromaffin cells that underwent neoplastic transformation.[
We present here a case of a pathologically diagnosed isolated pituitary NET, which underwent endoscopic endonasal transsphenoidal subtotal resection. Serial prospective and extensive metastatic workup revealed no other primary lesions during a 4-year follow-up period, indicating this to be the primary site of involvement. Finally, clinical and pathological aspects, imaging findings, surgical strategies, and the outcome of NET are discussed in the light of the pertinent literature.
CASE DESCRIPTION
A 65-year-old woman was admitted to our department following a 2-week history of rapidly progressive visual reduction, drowsiness, polyuria, and polydipsia. Her medical history included a temporal arachnoid cyst, symptomatic with seizures, and VIII cranial nerve schwannoma treated with gamma-knife radiosurgery 2 years before. Her clinical examination revealed no neurological deficit other than bitemporal hemianopsia; there was no diarrhea or flushing. Magnetic resonance imaging (MRI) showed a sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal compressing the optic chiasm and extending laterally toward the left cavernous sinus. The lesion was predominantly isointense to the gray matter on T1-weighted images (WI) and hyperintense on T2-WI [
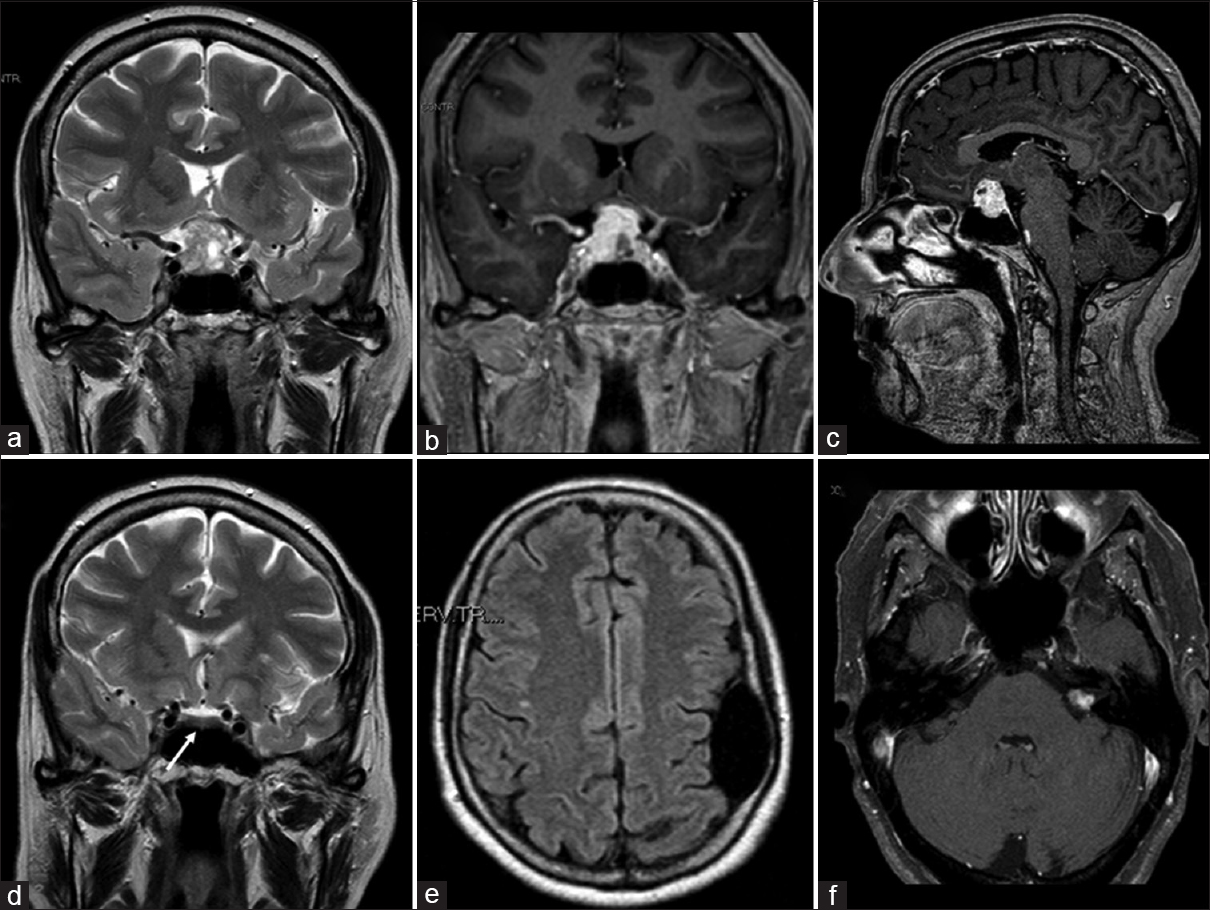
Figure 1
(a) Preoperative magnetic resonance (MRI) coronal T2-weighted imaging demonstrating sellar/suprasellar mass (diameter of 2 cm) with a heterogeneous signal, mainly hyperintense, compressing the optic chiasm and extended laterally toward the left cavernous sinus. (b and c) Preoperative magnetic resonance coronal and sagittal contrast-enhanced T1-weighted imaging showing an intense and diffuse enhancement. (d-f) MRI imaging performed 2 years before showing normal pituitary/sellar signal (white arrow), the frontal arachnoid cyst and left vestibular schwannoma
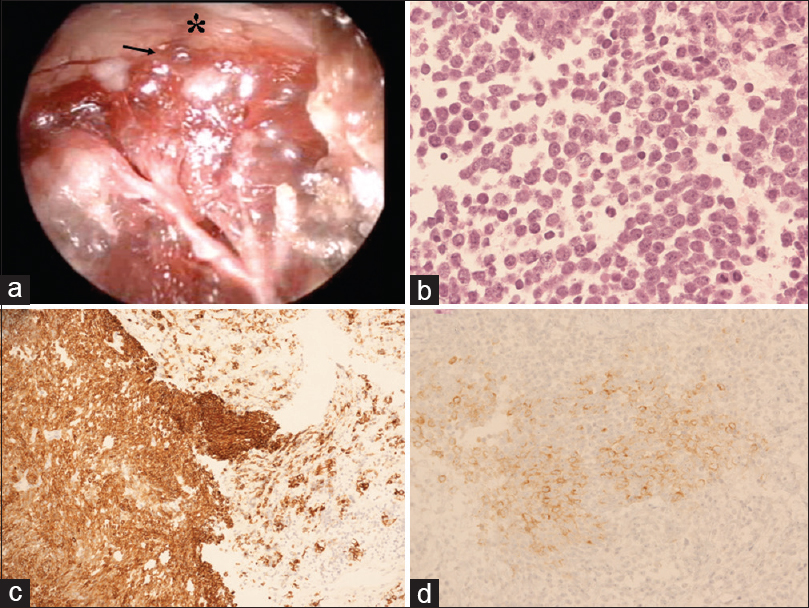
Figure 2
(a) Intraoperative endoscopic picture depicting that the upper part of the lesion (black arrow) presented clear infiltration of the optic chiasm (asterisk) without a safe plane of cleavage. (b) Hematoxylin-eosin (H and E) staining demonstrates cells with scant cytoplasm and nuclei with speckled chromatin. Mitotic activity was present and vigorous (magnification ×20). (c and d) Cells are positive for (c) cytokeratin Cam 5.2. (magnification, ×20) and (d) synaptophysin (magnification, ×20)
Histological and immunohistochemical analysis of the pituitary biopsy showed a neuroendocrine neoplasm of at least intermediate grade. The pituitary mass was characterized by cells with scant cytoplasm and nuclei with speckled chromatin. Mitotic activity was present and vigorous (up to 9–10 mitoses per high power field). There was no evidence of necrosis [
The postoperative course was uneventful, and neurological examination revealed a complete visual recovery. Before the patient was discharged, an extensive search for a primary carcinoid tumor or other metastases was undertaken. Images from CT of the thorax and abdomen were normal. Both 11C-5- hydroxytryptophan PET and Somatostatin Receptor scintigraphy using Indium 111Pentetreotide [111In-DTPA-D-Phe-] (Octreoscan) showed no additional area of abnormal uptake; therefore, primary pituitary NET was confirmed.
Subsequent postoperative CT and MRI [
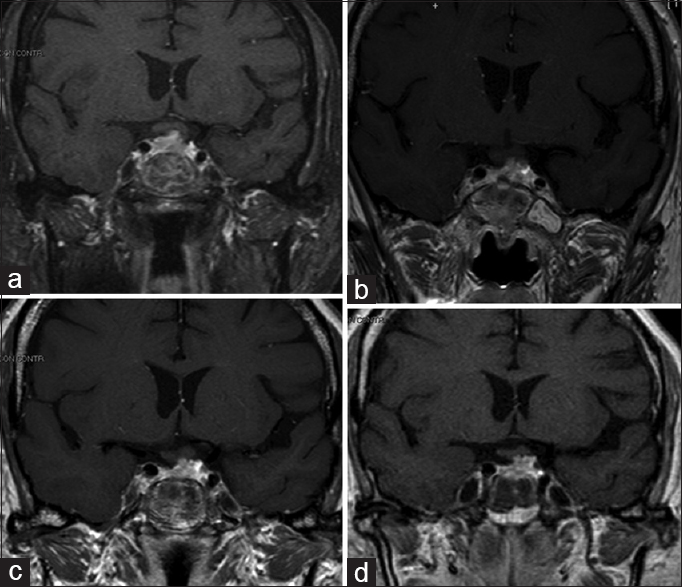
Fractioned stereotactic radiotherapy (SRT), employing image integration techniques and a frame that could be relocated to facilitate a fractioned dosing scheme, was carried out under a plan for reducing the treatment risk of the optic apparatus. Postoperative CT and MRI imaging were employed for greater precision in identifying the target. The relevant anatomy and tumor residual volume were then outlined. Because the tumor was close to critical normal tissue such as the optic chiasm, optic nerves, and cavernous sinus, the rotational angles were planned to reduce the doses to these critical regions. The treatment consisted of 8 daily fractions of 3.5 Gy to the 65% isodose line and a total dose of 43.1 Gy. Thus, the optic chiasm was exposed to a mean total dose of 208.6 cGy. No deterioration of visual acuity or pituitary function occurred after SRT.
Follow-up MRI studies at 12, 24, and 48 months were performed [Figure
DISCUSSION
We describe the treatment of a NET arising from enterochromaffin cells that had undergone neoplastic transformation.[
Metastatic carcinoid tumors in the central nervous system are an unusual but well-documented finding in several case reports.[
Patients with NET commonly present with no specific clinical features, including focal neurological deficit and intracranial hypertension. Functional tumors secreting one or more hormones would result in the presence of some endocrinal symptoms, and nonfunctional tumors may affect pituitary function and lead to hypopituitarism.[
Producing images of NET is usually challenging, requiring a combination of functional and anatomical techniques.[
Histopathological diagnosis is based on morphological and immunohistochemical examination. The neoplasm comprises cells of different sizes with uniform nuclei and scant cytoplasm; in addition, nidulant, basophil granulocytes are found. Irregular mitosis is present based on the degree of differentiation. The diagnosis of carcinoid tumor was favored based on its immunohistochemical profile; CAM 5.2, a marker for low molecular weight cytokeratins, synaptophysin, and chromogranin are markers for cells of neuroendocrine origin, and supported the diagnosis of NET.[
To date, only 6 cases of primary intracranial NET have been reported in the literature [
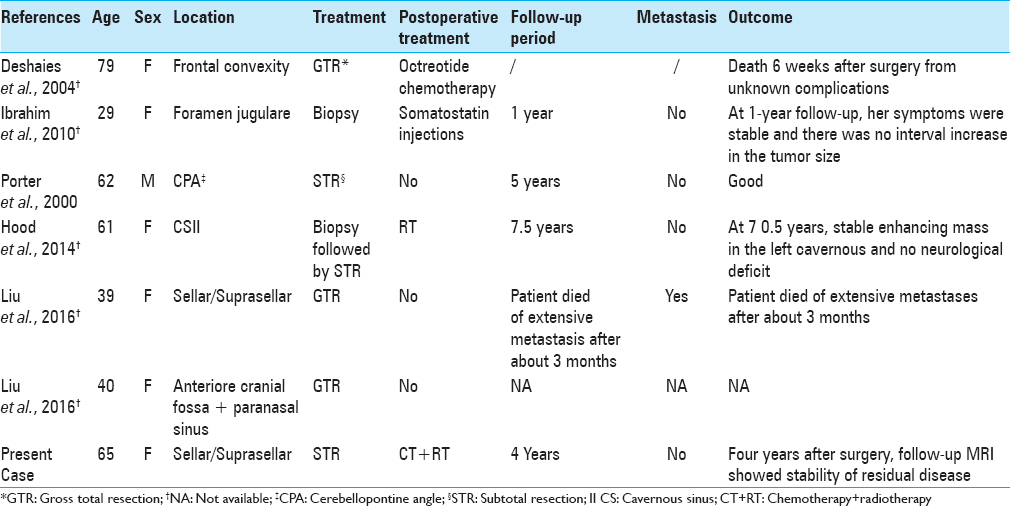
Ibrahim et al.[
Finally, Liu et al.[
Our case was the only one successfully treated with a multimodality approach. After a subtotal resection, the patient underwent to fractioned stereotactic radiotherapy (total irradiation dose 43.1 Gy, fractioned in 8 daily sessions), followed by polychemotherapy protocol (using a combination of cisplatinum, ifosfamide, and etoposide), which was well tolerated. Four years after the treatment, a follow-up MRI showed a complete stability of the residual disease and results from neurological examination were negative. Other particular features of our case were the location of NET and the use of an endoscopic technique. In fact, to the best of our knowledge, we present the second case of primary sellar NET. In contrast to the sellar NET presented by Liu et al.,[
CONCLUSIONS
Primary pituitary NET, though rare, should be included in the differential diagnosis of sellar lesions. A multimodality treatment approach is needed. Finally, the present case highlights that, in the case of a pituitary lesion infiltrating the optic chiasm, including NET, the endoscopic endonasal transsphenoidal subtotal resection followed by fractioned stereotactic radiotherapy and chemotherapy may represent an effective and safe choice of treatment.
Compliance with ethical standards.
Funding
No funding was received for this research.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Deshaies EM, Adamo MA, Qian J, DiRisio DA. A carcinoid tumor mimicking an isolated intracranial meningioma. Case report. J Neurosurg. 2004. 101: 858-60
2. Faggiano A, Mansueto G, Ferolla P, Milone F, del Basso de Caro ML, Lombardi G. Diagnosis and prognostic implication of the World Health Organization classification of neuroendocrine tumors. Endocrinol Invest. 2008. 31: 216-23
3. Hood B, Bray E, Bregy A, Norenberg M, Weed D, Morcos JJ. Primary carcinoid tumor of the cavernous sinus. World Neurosurg. 2014. 81: 202.e9-13
4. Ibrahim M, Yousef M, Bohnen N, Eisbruch A, Parmar H. Primary carcinoid tumor of the skull base: Case report and review of the literature. J Neuroimaging. 2010. 20: 390-2
5. Liu H, Wang H, Qi X, Yu C. Primary intracranial neuroendocrine tumor: Two case reports. World J Surg Oncol. 2016. 14: 138-
6. Patchell R, Posner J. Neurologic complications of carcinoid. Neurology. 1986. 36: 745-9
7. Porter DG, Chakrabarty A, McEvoy A, Bradford R. Intracranial carcinoid without evidence of extracranial disease. Neuropathol Appl Neurobiol. 2000. 26: 298-300
8. Sirsath NT, Babu KG, Das U, Premlatha CS. Paranasal sinus neuroendocrine carcinoma: A case report and review of the literature. Case Rep Oncol Med 2013. 2013. p.


