

- Department of Neurosurgery, Hospital de Pediatría Juan P. Garrahan, Ciudad Autónoma de Buenos Aires, Argentina
- Department of Oncology, St. Jude Children’s Research Hospital, Memphis, United States.
Correspondence Address:
Ramiro José del Río, Department of Neurosurgery, Hospital de Pediatría Juan P. Garrahan, Ciudad Autónoma de Buenos Aires, Argentina.
DOI:10.25259/SNI_681_2023
Copyright: © 2024 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Ramiro José del Río1, Santiago Ezequiel Cicutti1, Daniel C. Moreira2, Javier Danilo Gonzalez Ramos1. New CNS tumor classification: The importance in pediatric neurosurgical practice. 12-Apr-2024;15:130
How to cite this URL: Ramiro José del Río1, Santiago Ezequiel Cicutti1, Daniel C. Moreira2, Javier Danilo Gonzalez Ramos1. New CNS tumor classification: The importance in pediatric neurosurgical practice. 12-Apr-2024;15:130. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=12852
Date of Submission
12-Aug-2023
Date of Acceptance
26-Mar-2024
Date of Web Publication
12-Apr-2024
Abstract
Background: The management of the central nervous system (CNS) tumors in the pediatric population is crucial in neurosurgical practice. The World Health Organization (WHO) has evolved its classification of CNS tumors from the 19th century to the 5th edition, published in 2021, incorporating molecular advancements. This transition from morphology to molecular characterization is ongoing.
Methods: This manuscript analyzes the modifications introduced in the 5th edition of WHO’s CNS tumor classification, particularly focusing on pediatric tumor families. The paper integrates clinical, morphological, and molecular information, aiming to guide pediatric neurosurgeons in their daily practice and interdisciplinary discussions.
Results: The 5th edition of the WHO classification introduces a hybrid taxonomy that incorporates both molecular and histological components. The terminology shifts from “entity” to “type” and “subtype,” aiming to standardize terminology. Tumor grading experiences changes, integrating molecular biomarkers for prognosis. The concept of integrated layered diagnosis is emphasized, where molecular and histological information is combined systematically.
Conclusion: The 5th edition of the WHO CNS classification signifies a paradigm shift toward molecular characterization. The incorporation of molecular advances, the layered diagnostic approach, and the inclusion of clinical, morphological, and molecular information aim to provide comprehensive insights into pediatric CNS tumors. This classification offers valuable guidance for pediatric neurosurgeons, aiding in precise diagnosis and treatment planning for these complex neoplasms.
Keywords: Brain tumors, Central nervous system, Pediatric central nervous system tumors, Pediatric neurooncology, Pediatrics, World Health Organization classification
INTRODUCTION
The management of the central nervous system (CNS) tumors in children and adolescents is a rapidly evolving field. Pediatric CNS tumors represent approximately 20% of the burden of childhood malignancies. For pediatric neurosurgeons, this group of diseases is of high importance to neurosurgical practice due to the importance of a comprehensive interdisciplinary approach.
In 1979, the World Health Organization (WHO) published the first classification of CNS tumors, attempting to group different entities based on their common histopathologic features. Over the years, as knowledge about this type of neoplasm has advanced, several updates have been made, leading up to the 5th edition published in 2021. The purpose of this report is to highlight the most important points about the most common tumors in the pediatric population, considering the newest classification and providing key information of interest to pediatric neurosurgeons.
HISTORICAL CONTEXT
Virchow compiled the initial report on brain tumor classification in the mid-19th century.[
In 1988, Daumas-Duport et al. introduced an alternative CNS tumor grading system known as the St. Anne-Mayo grading scheme.[
The emergence of molecular biology, coupled with the reconsideration of the Zülch grading model, led to a revision of the fourth classification, published in 2016.[
Simultaneously, the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW), an entity distinct from the WHO, issued specific recommendations that were considered during the compilation of the new edition.[
In this manner, in 2021, building on the 2016 review and incorporating the latest advancements in clinical, morphological, and molecular domains, the 5th edition of the WHO CNS classification was released.
GENERAL CONSIDERATIONS AND MODIFICATIONS
The new classification takes into account both molecular and histological components, leading to a hybrid taxonomy that indicates an ongoing process of modification. The term “entity” has been replaced by “type,” and the term “subtype” is now used instead of “variant,” with the aim of standardizing nomenclature across diseases. Neoplasm nomenclature aims for simplicity by defining localization, age of presentation, and/or relevant genetic modifiers. For example, “diffuse midline glioma K27-altered” is used. Modifier terms such as “anaplastic” or “multiform” have been eliminated. Tumor grading has also changed, attempting to resemble more closely the grading of neoplasms outside the CNS while still preserving some classical features. As an illustration, Roman numerals have been substituted with Arabic numerals.[
Molecular biomarkers can serve as important prognostic indicators, which is why they have also been included in the determination of certain tumor grades. The Not Otherwise Specified (NOS) suffix, which groups tumors for which it is not possible to assign a specific category, is currently divided into two parts. On the one hand, the term “NOS” is still used for those entities that cannot be classified according to the WHO criteria due to a lack of specific tests, whether due to material shortages, sample impairment, or the absence of required molecular testing methods. On the other hand, the term “Not Elsewhere Classified” (NEC) is being added for those neoplasms where the necessary exams have been conducted, but the information is uncertain at the time of categorizing the sample into a specific type established by the WHO.[
INTEGRATED LAYER DIAGNOSTIC
Since the 2016 review, on the basis of a successful application for hematopoietic malignancies,[
This systematic approach serves as a guideline for pathologists when composing their reports. The concept involves a series of sequential evaluation levels. The first layer indicates the integrated diagnosis, the second identifies the histological type, and the third and fourth layers encompass the molecular features. From this model emerges the premise of “molecular biology defeats histology indicating a paradigm shift in tumor classification where molecular characteristics are increasingly prioritized over traditional histological features in determining diagnosis and prognosis.”[

2021 CLASSIFICATION OF PEDIATRIC TUMORS
As stated below, we will describe the most important changes that concern CNS tumor groups in childhood.
Gliomas
The first edition of the WHO classification has adopted a new approach to categorising glial, glioneuronal, and neural tumors. These tumors are now divided into six distinct families: (1) adult-type diffuse gliomas, (2) pediatric-type diffuse low-grade gliomas, (3) pediatric-type diffuse high-grade gliomas, (4) circumscribed astrocytic gliomas, (5) neuronal and glioneuronal tumors, and (6) ependymomas. Choroid plexus tumors are considered separately from these categories.[
Within this classification, a significant distinction between pediatric and adult gliomas becomes evident. While they may exhibit similar histology, there are well-known biological differences between them. An example of this distinction is that pediatric gliomas rarely transform higher histologic grades.
Pediatric diffuse low-grade gliomas comprise four distinct types: Diffuse astrocytoma MYB or MYBL1 altered, angiocentric glioma, polymorphous low-grade neuroepithelial tumor of the young, and diffuse low-grade glioma MAPK pathway altered [

There are also four types of diffuse high-grade gliomas: H3 K27-altered diffuse midline glioma, H3 G34 midline diffuse hemispheric glioma, pediatric-type diffuse high-grade glioma with H3 wild type and isocitrate dehydrogenase wild type, and infant-type hemispheric glioma. These types represent 10% of brain tumors in children and are associated with a very poor prognosis.[
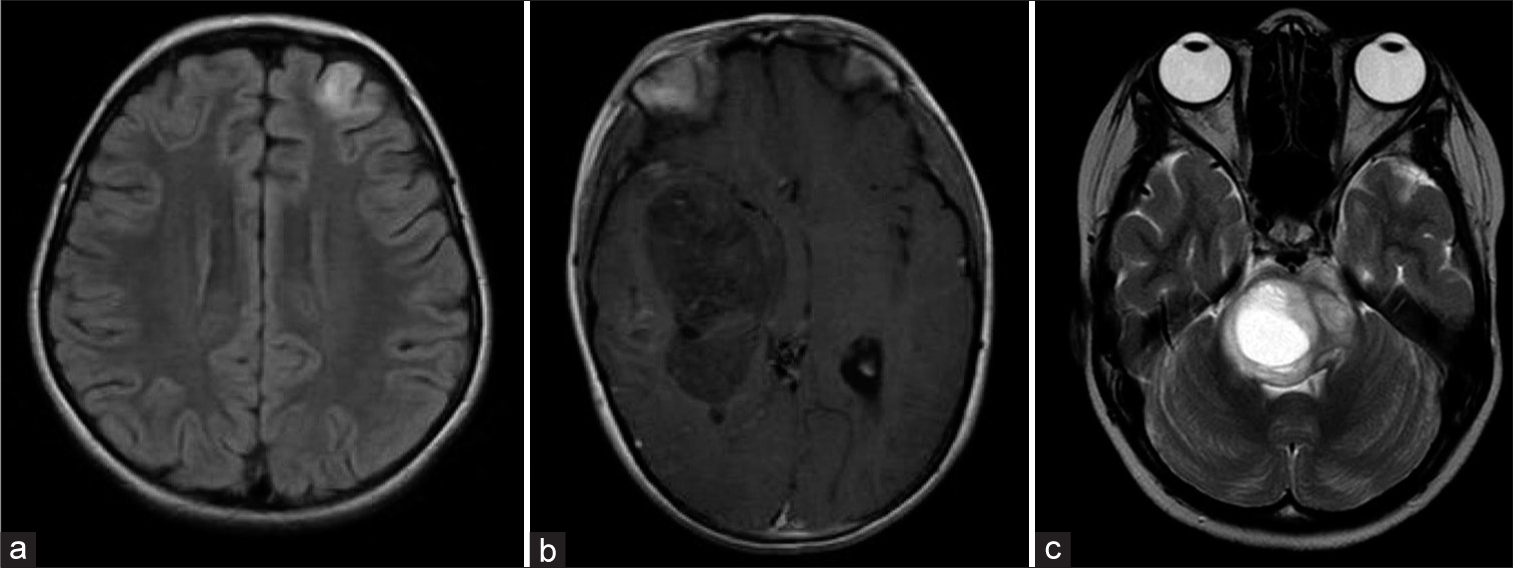
Figure 2:
Pediatric-type diffuse gliomas: (a) MRI FLAIR, axial section of a 7-year-old patient with an angiocentric glioma (pediatric-type diffuse low-grade glioma), grade 1. (b) MRI T1 with gadolinium, axial section of a 12-year-old patient with a diffuse hemispheric glioma H3 G34-mutant, grade 4 (formerly glioblastoma). (c) MRI T2 axial section of a 7-year-old patient with diffuse midline glioma H3 K27-altered, grade 4 (former diffuse intrinsic pontine glioma). MRI: Magnetic resonance imaging, FLAIR: Fluid attenuated inversion recovery.
In addition, it is important to note the disappearance of two types in pediatrics, oligodendroglioma and glioblastoma, tumors that pertain to the adult population.
Circumscribed astrocytic gliomas differ from the previous ones by being well-defined solid lesions.[
Another common tumor within this family in children is the pleomorphic xanthoastrocytoma (PXA), which can be either grade 2 or 3 and stands as one of the few examples of tumoral progression [

Figure 3:
Circumscribed astrocytic gliomas: (a) MRI T1 with gadolinium, axial section of a 2-year-old patient suffering from a posterior fossa pilocytic astrocytoma, WHO grade 1. (b) MRI T1 with gadolinium, axial section of a 1-year-old patient with a suprasellar pilocytic astrocytoma, WHO grade 1. (c) MRI T2, axial section of a 13-year-old patient with pleomorphic xanthoastrocytoma, WHO grade 3 (former anaplastic pleomorphic xanthoastrocytoma). MRI: Magnetic resonance imaging, WHO: World Health Organization.
Neural and neuronal-glial tumors
Tumors with a neural component are grouped into the new classification. To the previously known, three new types were added (even though the first one is still provisional): DGONC, myxoid glioneuronal tumor, and multinodular and vacuolating neuronal tumor [
Ependymomas
Ependymomas are third in terms of frequency among children, after gliomas and medulloblastomas (comprising 5–10% of cases). Around 90% of ependymomas are intracranial, with the majority arising in the posterior fossa (PF).[

Figure 4:
Classification of ependymomas according to the WHO classification 2021: This tumor type is categorized based on both its histological location and molecular characteristics. WHO: World Health Organization. ZFTA: Zinc finger translocation associated (previously known as C11orf95), YAP: Yes-associated protein, PF: Posterior Fossa, MYCN: Myelocytomatosis viral oncogene neuroblastoma derived homolog.

Figure 5:
Ependymomas: (a) MRI T2, Axial section. 2-year-old patient with ependymoma PFA, WHO grade 3. (b) MRI T2, Axial section of a 2-year-old patient with a supratentorial ependymoma ZFTA fusion-positive, WHO grade 3. (c) MRI T1 with gadolinium, Sagittal section. 12-year-old patient with a spinal myxopapillary ependymoma, WHO grade 2. MRI: Magnetic resonance imaging, WHO: World Health Organization, PFA: Posterior fossa type A. ZFTA: Zinc finger translocation associated (previously known as C11orf95).
Supratentorial ependymomas are classified based on two molecular fusions. On one hand, the C11orf95-RELA fusion is present in 70% of cases. At present, it is referred to as ZFTA (C11orf95 gene designation), given that it might fuse with more ligands than RELA.[
In relation to PF ependymomas, the division has ultimately been incorporated based on the methylation profile into two more common subtypes (A and B). Subtype A (PFA) is characterized by a relative loss of the epigenetic marker trimethylation H3K27 and is associated with a worse outcome. Subtype B (PFB) is more prevalent in older children, and although it is associated with better survival, its prognostic value is not significant in patients who have received conformal radiotherapy.[
Choroid plexus tumors
The main modification observed is the grouping of these types of neoplasms into one family. They have been separated from neuroepithelial primary tumors. The remaining classification of choroid plexus neoplastic tumors has not undergone significant changes.[
Embryonal tumors
Embryonal tumors constitute a heterogeneous type of malignant neoplasm of the CNS.[
In 2016, these types of entities were reclassified according to molecular profiles combined with histological features. In this way, the term PNET disappeared, grouping them in a unique family of CNS embryonal tumors. Based on an integrated taxonomy and putting special emphasis on molecular profile, the WHO 5th edition associates two groups: medulloblastomas and other embryonal tumors [

Medulloblastoma
Medulloblastomas constitute the most frequent solid malignant tumor in pediatrics.[
Factors related to poor outcomes included dissemination at the time of presentation, young age (<3 or five years), and residual tumor after surgery (>1.5 cm).[

Even though there is a correlation between morphological and molecular variants, the new classification may inform them according to the analyzed features: medulloblastoma histologically defined or medulloblastoma genetically defined.[
Other embryonal tumors
The rest of the embryonal tumors include the following types: Atypical teratoid rhabdoid tumors, embryonal tumor with multilayered rosettes, CNS neuroblastoma, FOXR2- activated, and CNS tumor with BCOR internal tandem duplication. While the first two were present in prior classifications, the last three have been included in the latest classification. There has been an important discussion about the incorporation of BCOR-altered tumors, considering their potential neuroectodermal nature due to features resembling mesenchymal neoplasms [

Figure 7:
Embryonal tumors: (a) MRI T2, an axial section of an 11-year-old boy with a histologically defined medulloblastoma (desmoplastic), WHO grade 4. (b) MRI T1 with gadolinium, an axial section of an 8-year-old patient with a molecularly and histologically defined hemispheric medulloblastoma. Subtype: desmoplastic nodular. SHH TP53-mutant. WHO grade 4. (c) MRI T2, an axial section of a 2-year-old patient with a CNS tumor with BCOR internal tandem duplications. WHO grade 4. MRI: Magnetic resonance imaging, WHO: World Health Organization, SHH: Sonic hedgehog, CNS: Central nervous system. BCOR: BCL6 corepressor.
Nevertheless, many times, it is not possible to count with all diagnostic methods to classify them, giving rise to being informed as NOS or NEC embryonal tumors if they do not have molecular features that could categorize them in some of the previously described.[
Pineal tumors
The types previously included are still present in this group: pineocytoma, pineal parenchymal tumor of intermediate differentiation, and pineoblastoma. The 2021 classification introduced the addition of the desmoplastic myxoid tumor of the pineal region SMARCB1-mutant, a rare neoplasm without histopathological signs of malignancy.[
Except for pinealocytomas and pinealoblastomas, the behavior of the remaining neoplasms is not completely understood, which makes it impossible to define their tumor grade.[
On the other hand, molecular groups based on methylation have been described for pinealoblastomas, showing different behaviors and prognoses. However, these groups were not included in the WHO 2021 classification.[
Craniopharyngioma
Craniopharyngioma constitutes a unique tumor with two variants: adamantinomatous and papillary. At present, they are classified as two different types of neoplasms due to their clinical, demographic differences, radiological features, histopathological findings, and molecular disorders.[
CONCLUSION
CNS tumor classifications have been described by several eminent professionals since the middle of the 19th century, resulting in the necessary publication by the WHO of a useful system to establish a common language worldwide. From 1979 up to the present day, five different editions have been released over the years.
The fifth edition consolidates the paradigm change thanks to molecular advances, even though the transition between morphological characterization and molecular biology is still in process. The advances, as described in the layer report from 2016, include the replacement of Roman numerals with Arabic ones, the exclusion of entities, the introduction of new family groups in the case of gliomas, and the description of tumor types and subtypes based on their molecular features. In addition, the incorporation of c-IMPACT-NOW reports marks a significant change, with special emphasis on neoplasms that affect younger patients.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent was not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Bailey P, Cushing H, editors. A classification of tumors of the glioma group on a histogenetic basis with a correlation study of prognosis. Philadelphia, PA: Lippincott and Williams; 1926. p.
2. Banan R, Hartmann C. The new WHO 2016 classification of brain tumors-what neurosurgeons need to know. Acta Neurochir (Wien). 2017. 159: 403-18
3. Becker AP, Scapulatempo-Neto C, Carloni AC, Paulino A, Sheren J, Aisner D. KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2015. 74: 743-54
4. Chatwin HV, Cruz Cruz J, Green AL. Pediatric high-grade glioma: Moving toward subtype-specific multimodal therapy. FEBS J. 2021. 288: 6127-41
5. Clarke M, Mackay A, Ismer B, Pickles J, Tatevossian R, Newman S. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020. 10: 942-63
6. Cohen A. Brain tumors in children. N Engl J Med. 2022. 386: 1922-31
7. Daumas-Duport C, Scheithauer BW, O'Fallon J, Kelly P. Grading of astrocytomas. A simple and reproducible method. Cancer. 1988. 62: 2152-65
8. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert M, Gilbertson R. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020. 30: 863-6
9. Guerreros Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019. 10: 4343
10. Holsken A, Sill M, Merkle J, Schweizer L, Buchfelder M, Flitsch J. Adamantinomatous and papillary craniopharyngiomas are characterized by distinct epigenomic as well as mutational and transcriptomic profiles. Acta Neuropathol Commun. 2016. 4: 20
11. Jones DT, Kocialkowski LL, Pearson D, Bäcklund LM, Ichimura K, Collins V. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008. 68: 8673-7
12. Kernohan JW, Mabon RF, Svien HJ, Adson AW. A simplified classification of the gliomas. Symposium on a new simplified concept of gliomas. Proc Staff Meet Mayo Clin. 1949. 24: 71-5
13. Kleihues P, Burger PC, Scheithauer BW, editors. Histological typing of tumors of the central nervous system. Heidelberg, New York: Springer Verlag; 1993. p.
14. Kleihues P, Cávenee WK, editors. Pathology and genetics of tumors of the nervous system. Lyon: IARC Press; 2000. p.
15. Kristensen BW, Priesterbach-Ackley LP, Petersen JK, Wesseling P. Molecular pathology of tumors of the central nervous system. Ann Oncol. 2019. 30: 1265-78
16. Li BK, Al-Karmi S, Huang A, Bouffet E. Pediatric embryonal brain tumors in the molecular era. Expert Rev Mol Diagn. 2020. 20: 293-303
17. Li BK, Vasiljevic A, Dufour C, Yao F, Ho BL, Lu M. Pineoblastoma segregates into molecular sub-groups with distinct clinico-pahtologic features: A Rare Brain Tumor Consortium registry study. Acta Neuropathol. 2020. 139: 223-41
18. Louis DN. The next step in brain tumor classification:” Let us now praise famous men”. or molecules? Acta Neuropathol. 2012. 124: 761-2
19. Louis DN, editors. Who classification of tumors of the central nervous system. Lyon: IARC Press; 2003. p.
20. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellision DW, Figarella-Branger D, editors. WHO classification and grading of tumors of the central nervous system. Lyon: IARC Press; 2016. p.
21. Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A. International Society of NeuropathologyHaarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014. 24: 429-35
22. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, FigarellaBranger D. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021. 23: 1231-51
23. Louis DN, Wesseling P, Aldape K, Brat D, Capper D, Cree IA. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020. 30: 844-56
24. Louis DN, Wesseling P, Paulus W, Giannini C, Batchelor T, Cairncross JG. cIMPACT-NOW update 1: Not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol. 2018. 135: 481-4
25. Marinoff AE, Ma C, Guo D, Snuderl M, Wright K, Manely P. Rethinking childhood ependymoma: A retrospective, multi-center analysis reveals poor long-term overall survival. J Neurooncol. 2017. 135: 201-11
26. McKean-Cowdin R, Razavi P, Barrington-Trimis J, Baldwin RT, Asgharzadeh S, Cockburn M. Trends in childhood brain tumor incidence, 1973-2009. J Neurooncol. 2013. 115: 153-60
27. Merchant TE, Bendel AE, Sabin ND, Burger P, Shaw D, Chang E. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Oncol. 2019. 37: 974-83
28. Millard NE, De Braganca KC. Medulloblastoma. J Child Neurol. 2016. 31: 1341-53
29. Mueller S, Chang S. Pediatric brain tumors: Current treatment strategies and future therapeutic approaches. Neurotherapeutics. 2009. 6: 570-86
30. Müller HL, Merchant TE, Warmuth-Metz M, Martinez-Barber JP, Puget S. Craniopharyngioma. Nat Rev Dis Primers. 2019. 5: 75
31. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. 2018. 20: iv1-86
32. Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: Clinical and biologic aspects. Neuro Oncol. 1999. 1: 232-50
33. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017. 134: 705-14
34. Paulina AC, Wen BC, Buatti JM, Hussey D, Zhen WK, Mayr NA. Intracranial ependymomas: An analysis of prognostic factors and patterns of failure. Am J Clin Oncol. 2002. 25: 117-22
35. Pfaff E, Aichmüller C, Sill M, Stichel D, Snuderl M, Karajannis MA. Molecular subgrouping of primary pineal parenchymal tumors reveals distinct subtypes correlated with clinical parameters and genetic alterations. Acta Neuropathol. 2020. 139: 243-57
36. Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983. 42: 1-15
37. Rutkowski S, von Hoff K, Emser A, Zwiener I, Pietsh T, Figarella-Branger D. Survival and prognostic factors of early childhood medulloblastoma: An international meta-analysis. J Clin Oncol. 2010. 28: 4961-8
38. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012. 16: 103-19
39. Sturm D, Pfister SM, Jones DT. Pediatric gliomas: Current concepts on diagnosis, biology, and clinical management. J Clin Oncol. 2017. 35: 2370-7
40. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, editors. WHO classification of tumors of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2008. p.
41. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford S. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012. 123: 465-72
42. Virchow R, editors. Die krankhaften geschwulste. Berlin: Hirschwald; 1863. p.
43. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics. CA Cancer J Clin. 2014. 64: 83-103
44. Wefers A, Bens S, Nemes K, Agaimy A, Oyen F, Vogelgesang S. Desmoplastic myxoid tumor, SMARCB1-mutant clinical, histopathological and molecular characterization of a pineal region tumor encountered in adolescents and adults. Acta Neuropathol. 2020. 139: 277-86
45. Wu J, Armstrong TS, Gilbert MR. Biology and management of ependymomas. Neuro Oncol. 2016. 18: 902-13
46. Zülch KJ, editors. Histological typing of tumors of the central nervous system. Geneva: World Health Organization; 1979. p.

