

- Department of Medicine, San Juan Bautista School of Medicine, Caguas, USA
- Neurologist, Caribbean Neurological Center, Caguas, USA
Correspondence Address:
Sara Zarei
Neurologist, Caribbean Neurological Center, Caguas, USA
DOI:10.4103/2152-7806.169561
Copyright: © 2015 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 16-Nov-2015;6:171
How to cite this URL: Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D, Orozco G, Chinea A. A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 16-Nov-2015;6:171. Available from: http://surgicalneurologyint.com/surgicalint_articles/a-comprehensive-review-of-amyotrophic-lateral-sclerosis/
Abstract
Amyotrophic lateral sclerosis (ALS) is a late-onset fatal neurodegenerative disease affecting motor neurons with an incidence of about 1/100,000. Most ALS cases are sporadic, but 5-10% of the cases are familial ALS. Both sporadic and familial ALS (FALS) are associated with degeneration of cortical and spinal motor neurons. The etiology of ALS remains unknown. However, mutations of superoxide dismutase 1 have been known as the most common cause of FALS. In this study, we provide a comprehensive review of ALS. We cover all aspects of the disease including epidemiology, comorbidities, environmental risk factor, molecular mechanism, genetic factors, symptoms, diagnostic, treatment, and even the available supplement and management of ALS. This will provide the reader with an advantage of receiving a broad range of information about the disease.
Keywords: Amyotrophic lateral sclerosis, sporadic and familial ALS, superoxide dismutase
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disorder that is, characterized by progressive loss of the upper and lower motor neurons (LMNs) at the spinal or bulbar level.[
ALS was first described in 1869 by French neurologist Jean-Martin Charcot.[
ALS is categorized in two forms. The most common form is sporadic (90–95%) which has no obvious genetically inherited component. The remaining 5–10% of the cases are familial-type ALS (FALS) due to their associated genetic dominant inheritance factor.[
Most of the reviews about ALS focus on a specific area of the diseases such as molecular mechanism, treatment, diagnostic, etc. This review will attempt to provide an up-to-date overview of all aspects of ALS. It will first cover the epidemiology and comorbidities of the disease, followed by known environmental risk factors such as smoking, chemical exposure, and radiation.
Improving the understanding of ALS pathogenesis is critical in developing earlier diagnostic methods as well as proposing new effective treatments. Thus, this review will present the most recent studies related to molecular mechanisms, genetics, ALS symptoms, diagnostic examinations, and treatments. Furthermore, due to the fact that there has been only one Food and Drug Administration (FDA) approved drug for ALS treatment, this review will also address nutritional supplements, as well as respiratory and nutritional managements that help alleviating the symptoms. This comprehensive study will inevitably lead to the better understanding of ALS and assist in extending the life expectancy associated with ALS by establishing a basis of knowledge that can be used to improve care.
THE EPIDEMIOLOGY OF AMYOTROPHIC LATERAL SCLEROSIS
During 1990's, the number of reported cases of ALS was between 1.5 and 2.7 per 100,000 in Europe and North America.[
The mean age of onset of ALS varies from 50 to 65 years with the median age of onset of 64 years old. Only 5% of the cases have an onset <30 years of age.[
Although most cases of ALS are sporadic, about 5% of the cases have a family history. The age of onset for FALS is about a decade earlier than for sporadic cases.[
A possible relationship between ALS and sports participation has been proposed but not demonstrated. In a cohort study of Italian professional football players, a severe increase in the incidence of ALS was found.[
ANALYSIS OF CO-MORBIDITIES OF AMYOTROPHIC LATERAL SCLEROSIS
Retrospective cohort studies have shown that the incidence in certain concomitant diseases and comorbidities is significantly different in ALS-affected population in comparison to the general population. German cohort studies examining comorbidities prior to diagnosis found that while cardiovascular risk factors were the most common comorbidities in ALS patients (31.5% vs. 40%), they still had a significantly higher incidence in the general population than in their affected cohort.[
Of the diseases most likely to be found in ALS patients prior to their diagnosis, a higher incidence in neurological disorders was noted when compared to the general population.[
Development of depression is one of the most common secondary symptoms associated with ALS. Previous studies have reported a prevalence of depression of 4–56% depending on the assessment measure.[
When comparing the survival rates of patients with mental health comorbidities, it was noted that patients suffering from Parkinson's and ALS had a slower progression of their disease and a greater survival time (P = 0.03).[
AMYOTROPHIC LATERAL SCLEROSIS ENVIRONMENTAL FACTORS
Previous epidemiologic studies have suggested that ALS patients may have been exposed to environmental toxins.[
SMOKING
Cigarette smoke has been found to increase the probability of developing ALS through inflammation, oxidative stress, and neurotoxicity by heavy metals contained in cigarettes.[
Physical activity
Athletes have higher ALS risk compared to the general population; however, performing passive to robust physical activity has not shown an increased susceptibility of developing ALS.[
Genetic profiles that promote physical fitness but not necessarily muscles strength could hold a proportional correlation between ALS and physical activity.[
Chemical exposure and metals
ALS has shown an association with exposure to agricultural chemicals such as pesticides, fertilizers, herbicides, insecticides, and formaldehyde.[
Among all the heavy metals that might be associated with ALS, lead exposure seems to be studied the most possibly due to the ALS-like symptoms experienced by people exposed to high concentrations of lead.[
It is thought that lead's role in ALS has to do with its ability to substitute for calcium in intracellular reactions leading to damage the mitochondria, oxidative damage to neurons, and strengthen glutamate's excitotoxicity.[
Radiation/electromagnetic fields
Laboratory studies have demonstrated that in vitro exposures to extremely lowfrequency electromagnetic waves generate a bigger quantity of cellular reactive oxygen than normal.[
Diet
Previous studies state that consuming high level of glutamate and fat can have adverse effects on ALS patients while Omega 3 fatty acids, Vitamin E, and fiber can have defensive impact.[
A large number of the information regarding environmental factors are based off questionnaires, all of which rely on subjects’ memories, leading to recall bias. Because of this, there may be a lack of information about the frequency and the amount of exposure to environmental factors. Also, this may also lead to the absence of biological markers in order to validate patient claims of exposure or pinpoint the possible action site.
Furthermore, due to ALS prolonged onset, it is difficult to isolate an exact environmental factor. In order to identify or narrow down possible ALS risk factors, a cohort study utilizing mice as a control and experimental group could be appropriate. Starting out with an emphasis on the most sought out factors (smoking, heavy metals, physical activity, diet, radiation, and chemical exposure). In order to track changes accurately, a type of biomarker specific to the possible risk factor could be designed, to theoretically track the progression of the disease.
MOLECULAR MECHANISM
Finding the molecular mechanisms by which motor neurons degenerate in ALS will aid in better understanding the disease's progress. Also, elucidation of molecular mechanisms can yield insight into developing strategies for newer treatments. The molecular basis of ALS is an intriguing issue that warrants in-depth research and investigation.
The most common cause of ALS is a mutation of the gene encoding the antioxidant enzyme superoxide dismutase 1 (SOD1).[
Glutamate excitotoxicity
Glutamate is synthesized in the presynaptic terminal. Uptake of glutamate into synaptic vesicles is facilitated by vesicular glutamate transporters.[
The motor cortex and spinal cord of ALS patients and transgenic SOD1 mouse model were found to have reduced astroglial glutamate transporter EAAT2, which leads to increased extracellular glutamate, over-stimulation of glutamate receptors, and excitotoxic neuronal degeneration.[
Aberrant EAAT2 messenger RNAs (mRNAs) were found in neuro-pathologically affected areas and cerebrospinal fluid (CSF) of ALS patients. These abnormalities included intron-retention and exon-skipping. According to these findings, aberrant mRNA is the main reason for the decrease in EAAT2 receptors among ALS patient.[
Structural and functional abnormalities of mitochondria
In addition to glutamate excitotoxicity, mitochondrial dysfunction also plays an important role in the motor neuron degeneration. Mitochondria are membrane bound organelles that have a significant role in vital processes such as intracellular energy production, cellular respiration, calcium homeostasis, and control of apoptosis.[
In the spinal cords of ALS patients, mutant SOD1 is deposited on the cytoplasmic face of the outer membrane and matrix of mitochondria.[
Mitochondria act as the powerhouse of every cell by converting energy into ATP that is, essential for the metabolism of the cells. Disturbed energy homeostasis and ATP deficits have been reported in the skeletal muscle biopsies of ALS patients. The normal process of electron transport chains is perturbed by the presence of mutant SOD1, causing less production of ATP. Some studies have demonstrated a decreased activity of respiratory chain complexes I and IV that are associated with defective energy metabolism.[
In addition to energy homeostasis, another major function of mitochondria in neurons regards buffering cytosolic calcium levels. Thus, unraveling the relationship between aberrant mitochondria, calcium dysregulation, and neuronal death is critical for the understanding of ALS pathogenesis. Calcium is one of the most significant intracellular messengers that play an important role in the regulation of metabolic pathways, neuronal development, and synaptic transmission. Mutant SOD1 has been found to disrupt calcium homeostasis. Several studies have shown that intracellular calcium is misregulated in ALS patients. A lower cytosolic Ca2+ buffering ability has been found as a principal risk factor for motor neuron damage.[
Several studies reported the loss of Ca2+ binding proteins such as calbindin-D28K and parvalbumin in the motor neurons of ALS patients.[
Regulating mitochondrial transport along axons is an essential task for the survival of neurons due to the mitochondria's key role in ATP generation, calcium buffering, and apoptotic signaling. Mitochondria are constantly being transported and docked at the same time in areas with high demand of ATP and calcium homeostasis such as growth cones, nodes of Ranvier, and synaptic terminals.[
Increased mitochondrial transport may slow axonal degeneration by delivering healthy mitochondria to axons while removing the damaged one from distal synapses.[
Finally, mutant SOD1 aggregates may also interfere with components of mitochondrial-dependent apoptotic machinery, such as B-cell lymphoma 2 (Bcl-2), which is a regulator protein that controls cell death.[
Impaired axonal structure and transport defects
Motor neurons are highly polarized cells with long axons that can be more than a meter in length and are thus vulnerable to damage. In addition to transmitting nerve impulses axons also transport organelles, RNA, proteins, lipids, and other cell parts to the axonal compartments. Moving toward the soma is called retrograde and is performed by cytoplasmic dynein molecular motors while moving toward the synaptic structures at the neuromuscular junction is an anterograde transport and is conducted by microtubule-dependent kinesin.[
Axonal transport in ALS patients is compromised. Dysregulation of axonal transport and the axonal compartment play a critical role in the pathophysiology of ALS. In several experiment with mutant SOD1 mice, loss of neurotrophic signaling and defective axonal transport were observed early in the disease process.[
Several pathways may be responsible for the impaired axonal transport in cases with mutant SOD1. Some of the most important mechanisms involve defective mitochondrial function or energy depletion, disruption of kinesin function by tumor necrosis factor, and excitotoxic damage by glutamate.[
Free radical-mediated oxidative stress
Reactive oxygen species (ROS) or free radicals form as natural byproducts of the normal metabolism of oxygen.[
Increased oxidative damage has been reported in ALS case biopsies and altered redox reactions were among the earliest theories of how mutant SOD1 could cause cytotoxicity.[
SOD1 is a major antioxidant protein, thus a mutation in this gene could cause cytotoxicity. Elevation of free radicals and increased oxidative damage were found in CSF, serum, and urine samples of ALS patients.[
GENETICS OF AMYOTROPHIC LATERAL SCLEROSIS
Sporadic ALS accounts for the majority of the cases of ALS, but genetic causes have been known to play a role.[
The clinical and pathological presentation of FALS and sporadic ALS are similar. Genetic testing can be used to differentiate inherited versus sporadic ALS and also to rule out other diseases that clinically mimic ALS.[
Starting with the discovery of mutations in the SOD1 gene, which codes for copper/zinc ion-binding SOD, 18 other genes have been identified in association with FALS.[
SOD1 mutations, which account for 20% of cases of FALS and 5% of SALS, cause cytotoxicity, which still has an unclear pathophysiology.[
FALS is inherited at a rate of 5–10% for all cases of ALS where family history of the disease is known.[
Close to 50% of FALS cases can be attributed to specific genes, and most are seemingly rare, highly penetrant, de novo mutations within affected families. Genome-wide association studies (GWAS) has allowed for the identification of common variables that are coupled to this disease.[
Family aggregation studies for SALS patients have shown that many people who have common neurodegenerative disorders also have ALS, possibly indicating the presence of a susceptible gene that could be responsible for increasing neurodegeneration in kindreds.[
Many GWAS for SALS have resulted in identifying genes that are associated to the ALS disease.[
AMYOTROPHIC LATERAL SCLEROSIS SYMPTOMS
The different ALS phenotypical expressions are classified mainly as: Limb-onset ALS with a combination of upper motor neuron (UMN) and LMN signs in the limbs; bulbar onset ALS, characterized with speech and swallowing difficulties followed by limb weakening in later stages of the disease; PLS with pure UMN involvement; and finally, PMA with pure LMN involvement.[
ALS patients experience localized muscle weakness that begins distally or proximally in their upper and lower limbs. Usually, the onset symptoms are asymmetric and develop in progressive generalized weakness and wasting of the muscles. The majority of the patients develop bulbar and respiratory symptoms and spasticity, which affects manual dexterity and gait.[
Speech disturbances tend to appear before the development of dysphagia for solids and liquids. Symptoms characteristic of limb-onset can develop simultaneously with bulbar symptoms occurring within 1–2 years. Patients with bulbar symptoms suffer from sialorrhea (excessive drooling) due to difficulty of swallowing saliva and minor bilateral lower facial weakness from UMN damage. The generalized weakness of the lower half of the face causes difficulty with lip seal and blowing cheeks.[
With the progression of ALS, patients develop the distinctive feature of a combination of upper motor and LMN degeneration signs within the same CNS region.[
Some uncommon symptoms of ALS include cramps and fasciculations in the absence of muscle weakness, and frontal lobe-type cognitive dysfunction.[
Weakness, spasticity, and abrupt deep tendon reflexes are usually characteristic of UMN disturbances involving the limbs. LMN features, on the other hand, include fasciculation, wasting of the muscle, and weakness. Spastic dysarthria characterized by slow, labored, and distorted speech is a consequence of bulbar UMN damage.[
In the majority of the cases, tendon reflexes become pathologically abrupt in a symmetrical pattern.[
In late stages of ALS, some patients develop flexor spasms or involuntary spams due to excess of activation of the flexor arc in spastic limbs.[
Other common symptoms in ALS are fatigue and reduced exercise capacity. As the disease progresses, patients require assistance with basic daily activities.[
The symptoms of ALS can be further divided into primary and secondary symptoms. Primary symptoms include muscle weakness and atrophy, spasticity, speech disturbances, poor management of oral secretions, difficulty swallowing, and respiratory complications that result in death. Secondary symptoms usually accompany primary symptoms, and they can significantly reduce the quality of life of patients, such as pain or difficulty performing daily tasks.[
Even though pain has not been associated with ALS, it has been reported in nearly 70% of ALS patients at some point during the course of the disease.[
Spasticity in ALS is usually due to changes in UMN within the motor cortex. Alteration in UMN processing can create the primitive reflex, also known as the Babinski sign, an important sign of neuropathy.[
DIAGNOSING AMYOTROPHIC LATERAL SCLEROSIS
The complexity and heterogeneous nature of ALS makes early and accurate diagnose a continuous challenge.[
Criteria and requirements for diagnosis
The El Escorial criteria for diagnosing ALS was published in 1994 by the World Federation of Neurology for inclusion standards for patients entering research studies and clinical trials.[
A definitive diagnosis of ALS requires evidence of LMN and UMN degeneration, and progression and spread of neurological symptoms or signs within or toward another anatomical region.[
Variability in clinical presentation
Based on the onset of symptoms, ALS is categorized as either a bulbar or spinal-onset disease, and further phenotypic subclassification is based on the extent of UMN and LMN dysfunction.[
Differential diagnosis
Lack of disease progression, an unusual patient history, or uncommon symptoms should prompt further investigation of the differential diagnosis of ALS [
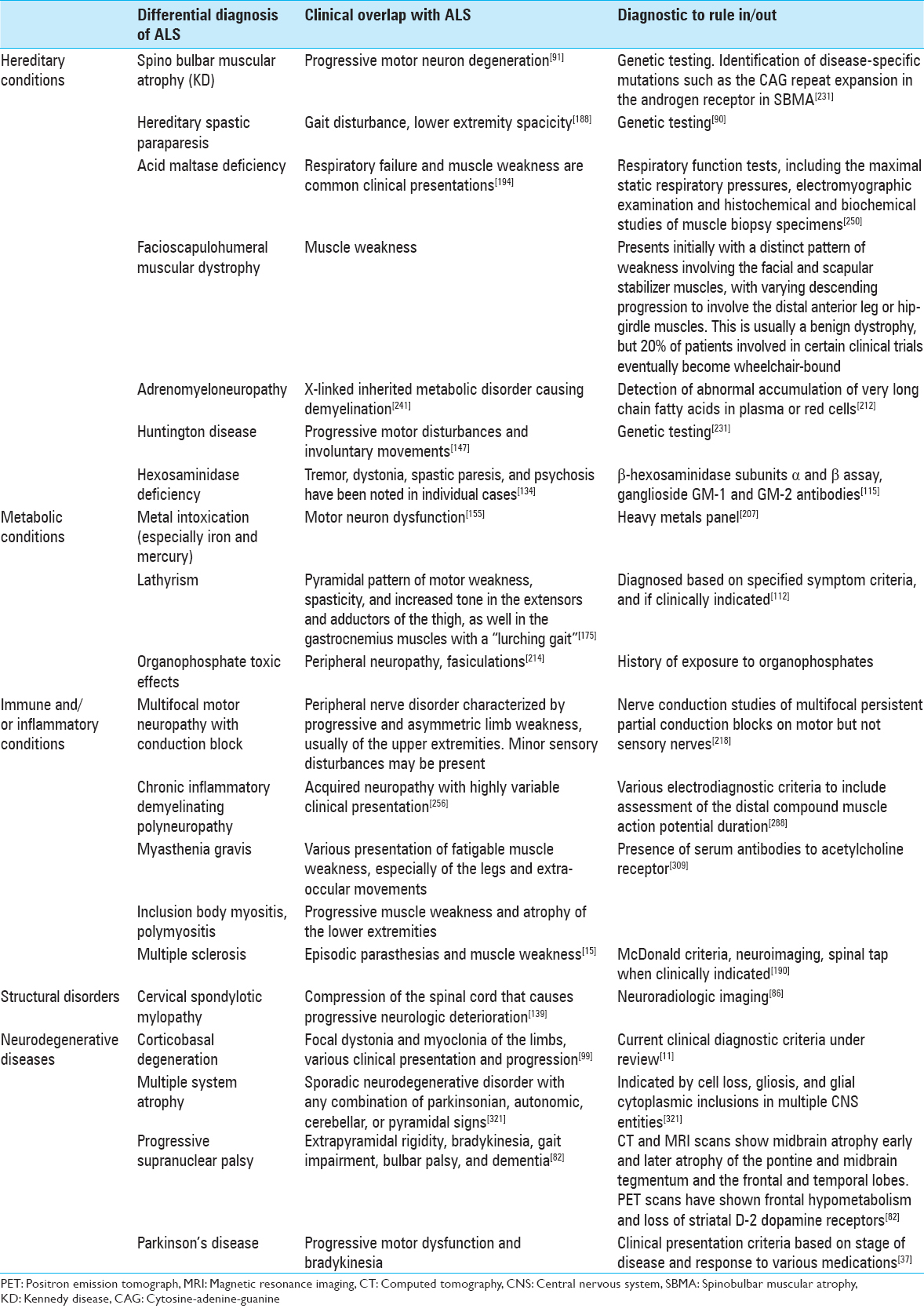
Common misdiagnosis of amyotrophic lateral sclerosis
Conditions that are commonly mistaken for or difficult to differentiate from ALS are multifocal motor neuropathy with conduction block, cervical spondylotic myelopathy, Kennedy disease (KD), and Post-polio syndrome (PPS).[
PPS presents with focal muscle weakness that very slowly progresses to other muscle groups over many years, and does not usually cause death.[
Diagnostic tests
There is no single or absolute test for ALS, but an extensive workup is done to help rule out the various differential diagnosis.

Electrodiagnostic tests
Electrodiagnostic studies are a useful diagnostic tool in the investigation of patients who may have ALS. EMG and nerve conduction studies are most sensitive to detecting the disease and can quantify its trademark characteristic of LMN degeneration.[
Laboratory studies
Typical labs drawn are erythrocyte sedimentation rate, serum and urine protein electrophoresis, thyroid function tests, serum calcium and phosphate measurements, and CSF analysis.[
Neuroimaging
Magnetic resonance imaging (MRI) studies of the brain and spinal cord are the most useful neuroimaging technique in ALS mainly to exclude syndromes that mimic ALS.[
TREATMENT
It has been suggested that there are shared environmental and genetic susceptibilities of several different neurological disorders, including PD, FTD, and ALS.[
Development of treatments to alleviate ALS symptoms is the foundation for providing proper healthcare to patients. In

Riluzole is currently the only FDA-approved drug treatment identified to have beneficial use in the survival of patients with ALS. Two clinical trials demonstrated evidence of increased survival for the riluzole-treatment group compared with controls.[
The recommendation dose of riluzole is 50 mg twice daily for patients with definite or probable ALS for duration <5 years, an FVC >60%, and no tracheostomy.[

AMYOTROPHIC LATERAL SCLEROSIS MANAGEMENT
Over the past two decades, the management of ALS has changed considerably. Although still incurable, ALS is not untreatable. Emphasis has been made in treatments and interventions that prolong survival.[
Multidisciplinary management
In recent years, multidisciplinary ALS treatment facilities have emerged as a result to a shift in the approach of health care delivery to ALS patients. By treating only ALS patients, these multidisciplinary ALS clinics gather great resources and clinical expertise that can facilitate the management and provide optimized care of this progressive disease.[
These multidisciplinary ALS specialized clinics can better assist in managing the complex issues associated with ALS, such as psychosocial problems, nutrition, dysphagia, dysarthria, functional decline, and respiratory symptoms. Both the American Academy of Neurology (AAN) and the European Federation of Neurological Societies recommended that after diagnosis, the patient and caregivers should be referred to a multidisciplinary clinic and receive regular support from a multidisciplinary care team to optimize health care delivery and prolong survival.[
Respiratory management
The most common cause of death in ALS is due to respiratory failure with or without pneumonia. The presenting symptoms of respiratory muscle weakness, secondary to progressive motor neuron degeneration, result in reduced ventilation.[
Since ALS mortality is mostly caused by respiratory failure, the assessment and management of respiratory function are of great importance. The most common and widely available measure for detecting respiratory decline is the examination of the patient's FVC.[
Nutritional management
Most ALS patients develop dysphagia which leads to malnutrition and weight loss. The consequences of this progressive deterioration include restricting ample nutrition, dehydration, choking, aspiration, and weight loss. As a consequence of the dysphagia present in these patients, the risk of insufficient caloric and fluid intake increases, leading to worsening of weakness and fatigue.[
DIETARY SUPPLEMENTATION
Vitamin E and Vitamin A
Although the pathophysiologic causes of ALS are not clearly understood, it is hypothesized that free radical stress is a main component of the cell degeneration contributing to ALS progression and onset.[
In other studies, the efficacy of Vitamin A (beta-carotene) supplementation was investigated among ALS patients. Their results showed that beta-carotene neither has any neuroprotective effect on ALS patients nor helps with slowing down the progression of the disease.[
Creatine
An investigational study by Klivenyi et al. on transgenic mouse with ALS showed a possible protective effect of dietary creatine supplementation on neurons.[
Pu-erh tea extract
A recent research study from Jilin University in China suggests that pu-erh tea extract (PTE) can help in preventing the rapid advancement of ALS in patients. The results of the study suggest that PTE can posttranscriptionally prevent the progression of FET family proteins that are associated with ALS. Also, results from the study suggest that PTE induces FUS/TLS protein degradation via lysosome-dependent pathway. With long-term intake, PTE may prevent protein aggregation and enable cells to maintain function within normal levels of protein. Further studies are required to ascertain the efficacy of PTE on FET in vivo.[
SURVIVAL AND PROGNOSIS
ALS is a progressive condition in which more than half of patients diagnosed do not survive within the first 30 months after symptom onset. Only 20% of the patients survive between 5 and 10 years after symptoms onset.[
Some ALS subtypes vary according to prognosis. LMN form of ALS, which includes flail-limb variant and PMA, shows a slower progression than other forms of ALS.[
CONCLUSION
This study covered a broad range of information about ALS from epidemiology to molecular mechanism and treatment of the disease. Unfortunately, ALS is considered an incurable disease, with an expected life expectancy of 3–5 years after the onset of symptoms.[
There have been important advancements in the understanding of ALS pathophysiology. Nineteen genes and genetic loci have been found that are associated with ALS.[
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abel EL. Football increases the risk for Lou Gehrig's disease, amyotrophic lateral sclerosis. Percept Mot Skills. 2007. 104: 1251-4
2. Abhinav K, Stanton B, Johnston C, Hardstaff J, Orrell RW, Howard R. Amyotrophic lateral sclerosis in South-East England: A population-based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry). Neuroepidemiology. 2007. 29: 44-8
3. Agosta F, Al-Chalabi A, Filippi M, Hardiman O, Kaji R, Meininger V. The El Escorial criteria: Strengths and weaknesses. Amyotroph Lateral Scler Frontotemporal Degener. 2015. 16: 1-7
4. Alavi A, Khani M, Nafissi S, Shamshiri H, Elahi E. An Iranian familial amyotrophic lateral sclerosis pedigree with p.Val48Phe causing mutation in SOD1: A genetic and clinical report. Iran J Basic Med Sci. 2014. 17: 735-9
5. Al-Chalabi A, Leigh PN. Trouble on the pitch: Are professional football players at increased risk of developing amyotrophic lateral sclerosis?. Brain. 2005. 128: 451-3
6. Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994. 36: 846-58
7. Al-exianu ME, Manole E, Engelhardt JI, Appel SH. Ultrastructural evidence of calcium involvement in experimental autoimmune gray matter disease. J Neurosci Res. 2000. 60: 98-105
8. AlSarraj S, King A, Cleveland M, Pradat PF, Corse A, Rothstein JD. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol Commun. 2014. 2: 165-
9. Andersen PM, Borasio GD, Dengler R, Hardiman O, Kollewe K, Leigh PN. Good practice in the management of amyotrophic lateral sclerosis: Clinical guidelines. An evidence-based review with good practice points. EALSC Working Group. Amyotroph Lateral Scler. 2007. 8: 195-213
10. Andreassen OA, Jenkins BG, Dedeoglu A, Ferrante KL, Bogdanov MB, Kaddurah-Daouk R. Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J Neurochem. 2001. 77: 383-90
11. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013. 80: 496-503
12. Atassi N, Cook A, Pineda CM, Yerramilli-Rao P, Pulley D, Cudkowicz M. Depression in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011. 12: 109-12
13. Azari MF, Profyris C, Le Grande MR, Lopes EC, Hirst J, Petratos S. Effects of intraperitoneal injection of Rofecoxib in a mouse model of ALS. Eur J Neurol. 2005. 12: 357-64
14. Barber SC, Shaw PJ. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010. 48: 629-41
15. Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain. 1997. 120: 2059-69
16. Beers DR, Ho BK, Siklós L, Alexianu ME, Mosier DR, Mohamed AH. Parvalbumin overexpression alters immunemediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem. 2001. 79: 499-509
17. Beghi E, Logroscino G, Chiò A, Hardiman O, Millul A, Mitchell D. Amyotrophic lateral sclerosis, physical exercise, trauma and sports: Results of a population-based pilot case-control study. Amyotroph Lateral Scler. 2010. 11: 289-92
18. Behnia M, Kelly JJ. Role of electromyography in amyotrophic lateral sclerosis. Muscle Nerve. 1991. 14: 1236-41
19. Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol. 2002. 249: 609-15
20. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994. 330: 585-91
21. Bernard-Marissal N, Moumen A, Sunyach C, Pellegrino C, Dudley K, Henderson CE. Reduced calreticulin levels link endoplasmic reticulum stress and Fast-riggered cell death in motoneurons vulnerable to ALS. J Neurosci. 2012. 32: 4901-12
22. Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo . Proc Natl Acad Sci U S A. 2010. 107: 20523-8
23. Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2010. 81: 639-45
24. Blatzheim K. Interdisciplinary palliative care, including massage, in treatment of amyotrophic lateral sclerosis. J Bodyw Mov Ther. 2009. 13: 328-35
25. Boillée S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron. 2006. 52: 39-59
26. Borasio GD, Miller RG. Clinical characteristics and management of ALS. Semin Neurol. 2001. 21: 155-66
27. Borasio GD, Voltz R, Miller RG. Palliative care in amyotrophic lateral sclerosis. Neurol Clin. 2001. 19: 829-47
28. Borasio GD, Voltz R. Palliative care in amyotrophic lateral sclerosis. J Neurol. 1997. 244: S11-7
29. Bordet T, Berna P, Abitbol JL, Pruss R. Olesoxime (TRO19622): A novel mitochondrial-targeted neuroprotective compound. Pharmaceuticals. 2010. 3: 345-68
30. Bordet T, Buisson B, Michaud M, Drouot C, Galéa P, Delaage P. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007. 322: 709-20
31. Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999. 46: 787-90
32. Boston-Howes W, Williams EO, Bogush A, Scolere M, Pasinelli P, Trotti D. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: Therapeutic implications for amyotrophic lateral sclerosis. Exp Neurol. 2008. 213: 229-37
33. Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011. 82: 196-203
34. Bradley WG, Mash DC. Beyond Guam: The cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph Lateral Scler. 2009. 10: 7-20
35. Brooks BR, Miller R, Swash M, Munsat TL. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2000. 1: 293-9
36. Brooks BR, Sanjak M, Roelke K. Phase 2B randomized dose ranging clinical trial of tamoxifen, a selective estrogen receptor modulator [SERM], in ALS: Sensitivity analyses of discordance between survival and functional outcomes with long-term follow-up. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005. 6: 118-
37. Brooks DJ. Parkinson's disease: Diagnosis. Parkinsonism Relat Disord. 2012. 18: S31-3
38. Busch MA, Maske UE, Ryl L, Schlack R, Hapke U. Prevalence of depressive symptoms and diagnosed depression among adults in Germany: results of the German Health Interview and Examination Survey for Adults. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2013. 56: 733-9
39. Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011. 82: 623-7
40. Carilho R, de Carvalho M, Kuehl U, Pinto S, Pinto A, Kromminga A. Erythropoietin and amyotrophic lateral sclerosis: Plasma level determination. Amyotroph Lateral Scler. 2011. 12: 439-43
41. Carrì MT, Valle C, Bozzo F, Cozzolino M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front Cell Neurosci. 2015. 9: 41-
42. Cats EA, van der Pol WL, Piepers S, Franssen H, Jacobs BC, van den Berg-Vos RM. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology. 2010. 75: 818-25
43. Chancellor AM, Slattery JM, Fraser H, Swingler RJ, Holloway SM, Warlow CP. The prognosis of adult-onset motor neuron disease: A prospective study based on the Scottish Motor Neuron Disease Register. J Neurol. 1993. 240: 339-46
44. Chang Y, Kong Q, Shan X, Tian G, Ilieva H, Cleveland DW. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One. 2008. 3: e2849-
45. Cheah BC, Kiernan MC. Dexpramipexole, the R(+) enantiomer of pramipexole, for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010. 13: 911-20
46. Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol. 2007. 166: 810-6
47. Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004. 74: 1128-35
48. Chiò A, Benzi G, Dossena M, Mutani R, Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005. 128: 472-6
49. Chiò A, Borghero G, Pugliatti M, Ticca A, Calvo A, Moglia C. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol. 2011. 68: 594-8
50. Chiò A, Bottacchi E, Buffa C, Mutani R, Mora G; PARALS. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J Neurol Neurosurg Psychiatry. 2006. 77: 948-50
51. Chiò A, Finocchiaro E, Meineri P, Bottacchi E, Schiffer D. Safety and factors related to survival after percutaneous endoscopic gastrostomy in ALS. ALS Percutaneous Endoscopic Gastrostomy Study Group. Neurology. 1999. 53: 1123-5
52. Chiò A, Gauthier A, Montuschi A, Calvo A, Di Vito N, Ghiglione P. A cross sectional study on determinants of quality of life in ALS. J Neurol Neurosurg Psychiatry. 2004. 75: 1597-601
53. Chung MJ, Suh YL. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol. 2002. 26: 3-7
54. Corcia P, Pradat PF, Salachas F, Bruneteau G, Forestier Nl, Seilhean D. Causes of death in a post-mortem series of ALS patients. Amyotroph Lateral Scler. 2008. 9: 59-62
55. Corcoran LJ, Mitchison TJ, Liu Q. A novel action of histone deacetylase inhibitors in a protein aggresome disease model. Curr Biol. 2004. 14: 488-92
56. Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet. 2010. 47: 190-4
57. Costa J, Swash M, de Carvalho M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis: A systematic review. Arch Neurol. 2012. 69: 1410-6
58. Cozzolino M, Rossi S, Mirra A, Carrì MT. Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front Cell Neurosci. 2015. 9: 31-
59. Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx. 2004. 1: 273-83
60. Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler. 2009. 10: 99-106
61. Cudkowicz ME, Shefner JM, Simpson E, Grasso D, Yu H, Zhang H. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008. 38: 837-44
62. Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006. 77: 390-2
63. Dangoumau A, Verschueren A, Hammouche E, Papon MA, Blasco H, Cherpi-Antar C. Novel SOD1 mutation p. V31A identified with a slowly progressive form of amyotrophic lateral sclerosis. Neurobiol Aging. 2014. 35: 266.e1-4
64. Davenport RJ, Swingler RJ, Chancellor AM, Warlow CP. Avoiding false positive diagnoses of motor neuron disease: Lessons from the Scottish Motor Neuron Disease Register. J Neurol Neurosurg Psychiatry. 1996. 60: 147-51
65. de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008. 119: 497-503
66. de Carvalho M, Matias T, Coelho F, Evangelista T, Pinto A, Luís ML. Motor neuron disease presenting with respiratory failure. J Neurol Sci. 1996. 139: 117-22
67. de Carvalho M, Pinto S, Costa J, Evangelista T, Ohana B, Pinto A. A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010. 11: 456-60
68. de Paulis T. ONO-2506.Ono. Curr Opin Investig Drugs. 2003. 4: 863-7
69. De Vos K, Severin F, Van Herreweghe F, Vancompernolle K, Goossens V, Hyman A. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J Cell Biol. 2000. 149: 1207-14
70. De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007. 16: 2720-8
71. Debono MW, Le Guern J, Canton T, Doble A, Pradier L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur J Pharmacol. 1993. 235: 283-9
72. Degens H, Alway SE. Control of muscle size during disuse, disease, and aging. Int J Sports Med. 2006. 27: 94-9
73. Deivasigamani S, Verma HK, Ueda R, Ratnaparkhi A, Ratnaparkhi GS. A genetic screen identifies Tor as an interactor of VAPB in a Drosophila model of amyotrophic lateral sclerosis. Biol Open. 2014. 3: 1127-38
74. Desnuelle C, Dib M, Garrel C, Favier A. A double-blind, placebocontrolled randomized clinical trial of alpha-tocopherol (Vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001. 2: 9-18
75. Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: Current status and future prospects. J Assoc Physicians India. 2004. 52: 794-804
76. Din JN, Newby DE, Flapan AD. Omega 3 fatty acids and cardiovascular disease – Fishing for a natural treatment. BMJ. 2004. 328: 30-5
77. Doblhammer G, Fink A, Fritze T. Short-term trends in dementia prevalence in Germany between the years 2007 and 2009. Alzheimers Dement. 2015. 11: 291-9
78. Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013. 80: 415-28
79. Dubner R, Gold M. The neurobiology of pain. Proc Natl Acad Sci U S A. 1999. 96: 7627-30
80. Duffy JR, Peach RK, Strand EA. Progressive apraxia of speech as a sign of motor neuron disease. Am J Speech Lang Pathol. 2007. 16: 198-208
81. Duffy JR. Apraxia of speech in degenerative neurologic disease. Aphasiology. 2006. 20: 511-27
82. Duvoisin RC, Golbe LI, Lepore FE. Progressive supranuclear palsy. Can J Neurol Sci. 1987. 14: 547-54
83. Echaniz-Laguna A, Zoll J, Ribera F, Tranchant C, Warter JM, Lonsdorfer J. Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann Neurol. 2002. 52: 623-7
84. Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) – Revised report of an EFNS task force. Eur J Neurol. 2012. 19: 360-75
85. Ellis CM, Dawson JM, Williams SC, Leigh PN. Distinct hyperintense MRI signal changes in the corticospinal tracts of a patient with motor neuron disease. Amyotroph Lateral Scler. 2000. 1: 41-4
86. Emery SE. Cervical spondylotic myelopathy: Diagnosis and treatment. J Am Acad Orthop Surg. 2001. 9: 376-88
87. Ferguson TA, Elman LB. Clinical presentation and diagnosis of amyotrophic lateral sclerosis. NeuroRehabilitation. 2007. 22: 409-16
88. Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011. 7: 616-30
89. Ferrante KL, Shefner J, Zhang H, Betensky R, O’Brien M, Yu H. Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology. 2005. 65: 1834-6
90. Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006. 6: 65-76
91. Fischbeck KH. Kennedy disease. J Inherit Metab Dis. 1997. 20: 152-8
92. Fitting JW, Paillex R, Hirt L, Aebischer P, Schluep M. Sniff nasal pressure: A sensitive respiratory test to assess progression of amyotrophic lateral sclerosis. Ann Neurol. 1999. 46: 887-93
93. Forbes RB, Colville S, Swingler RJ. The epidemiology of amyotrophic lateral sclerosis (ALS/MND) in people aged 80 or over. Age Ageing. 2004. 33: 131-4
94. Forsberg K, Andersen PM, Marklund SL, Brännström T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011. 121: 623-34
95. Ganzini L, Johnston WS, Hoffman WF. Correlates of suffering in amyotrophic lateral sclerosis. Neurology. 1999. 52: 1434-40
96. Garber K. Genetics. The elusive ALS genes. Science. 2008. 319: 20-
97. Geevasinga N, Menon P, Yiannikas C, Kiernan MC, Vucic S. Diagnostic utility of cortical excitability studies in amyotrophic lateral sclerosis. Eur J Neurol. 2014. 21: 1451-7
98. Ghadge GD, Slusher BS, Bodner A, Canto MD, Wozniak K, Thomas AG. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc Natl Acad Sci U S A. 2003. 100: 9554-9
99. Gibb WR, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain. 1989. 112: 1171-92
100. Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012. 11: 54-65
101. Goetz CG. Amyotrophic lateral sclerosis: Early contributions of Jean-Martin Charcot. Muscle Nerve. 2000. 23: 336-43
102. Goldstein LH, Atkins L, Landau S, Brown RG, Leigh PN. Longitudinal predictors of psychological distress and self-esteem in people with ALS. Neurology. 2006. 67: 1652-8
103. Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H. The natural history of primary lateral sclerosis. Neurology. 2006. 66: 647-53
104. Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007. 6: 1045-53
105. Gößwald A, Schienkiewitz A, Nowossadeck E, Busch MA. Prevalence of myocardial infarction and coronary heart disease in adults aged 40 to 79 years in Germany. Bundesgesundheitsblatt Health. 2013. 56: 650-655
106. Graf M, Ecker D, Horowski R, Kramer B, Riederer P, Gerlach M. High dose Vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: Results of a placebo-controlled double-blind study. J Neural Transm. 2005. 112: 649-60
107. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006. 38: 411-3
108. Groeneveld GJ, Veldink JH, van der Tweel I, Kalmijn S, Beijer C, de Visser M. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol. 2003. 53: 437-45
109. Gross TS, Poliachik SL, Prasad J, Bain SD. The effect of muscle dysfunction on bone mass and morphology. J Musculoskelet Neuronal Interact. 2010. 10: 25-34
110. Gruis KL, Lechtzin N. Respiratory therapies for amyotrophic lateral sclerosis: A primer. Muscle Nerve. 2012. 46: 313-31
111. Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK. Benefit of Vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996. 39: 147-57
112. Haimanot RT, Kidane Y, Wuhib E, Kalissa A, Alemu T, Zein ZA. Lathyrism in rural northwestern Ethiopia: A highly prevalent neurotoxic disorder. Int J Epidemiol. 1990. 19: 664-72
113. Hammer EM, Häcker S, Hautzinger M, Meyer TD, Kübler A. Validity of the ALS-Depression-Inventory (ADI-12) – A new screening instrument for depressive disorders in patients with amyotrophic lateral sclerosis. J Affect Disord. 2008. 109: 213-9
114. Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: A neglected aspect of disease. Neurol Res Int 2011. 2011. p.
115. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011. 7: 639-49
116. Havins W.editors. Apathy, Depression, and Emotional Lability in Patients with Amyotrophic Lateral Sclerosis. Houston, Texas: Diss University of Houston; 2014. p.
117. Hayashi H, Kato S. Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J Neurol Sci. 1989. 93: 19-35
118. He J, Mangelsdorf M, Fan D, Bartlett P, Brown MA. Amyotrophic lateral sclerosis genetic studies: From genomewide association mapping to genome sequencing. Neuroscientist. 2014. p.
119. Henriques A, Pitzer C, Schneider A. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand?. Front Neurosci. 2010. 4: 32-
120. Honig LS, Chambliss DD, Bigio EH, Carroll SL, Elliott JL. Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology. 2000. 55: 1082-8
121. Hortobágyi T, Cairns NJ.editors. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neuropathology of Neurodegenerative Diseases Book and Online. Cambridge University Press; 2014. p. 209-
122. Huang Y, Wu Z, Zhou B. HSOD1 promotes tau phosphorylation and toxicity in the Drosophila model. J Alzheimers Dis. 2015. 45: 235-44
123. Huisman MH, de Jong SW, van Doormaal PT, Weinreich SS, Schelhaas HJ, van der Kooi AJ. Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry. 2011. 82: 1165-70
124. Ikenaka K, Katsuno M, Kawai K, Ishigaki S, Tanaka F, Sobue G. Disruption of axonal transport in motor neuron diseases. Int J Mol Sci. 2012. 13: 1225-38
125. Ikiz B.editors. Unraveling the Molecular Mechanism Underlying ALS-Linked Astrocyte Toxicity for Motor Neurons. PhD diss. Columbia University; 2013. p.
126. Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009. 187: 761-72
127. Ilsemann J, Kollewe K, Dengler R, Krampfl K, Petri S. Analysis of co-morbidities in a database of ALS patients. Aktuelle Neurol. 2008. 35: P414-
128. Ilzecka J, Stelmasiak Z, Solski J, Wawrzycki S, Szpetnar M. Plasma amino acids concentration in amyotrophic lateral sclerosis patients. Amino Acids. 2003. 25: 69-73
129. Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014. 111: 197-201
130. Iwasaki Y, Ikeda K, Kinoshita M. Molecular and cellular mechanism of glutamate receptors in relation to amyotrophic lateral sclerosis. Curr Drug Targets CNS Neurol Disord. 2002. 1: 511-8
131. Jaiswal MK. Calcium, mitochondria, and the pathogenesis of ALS: The good, the bad, and the ugly. Front Cell Neurosci. 2013. 7: 199-
132. Jaiswal MK. Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: Implications for motoneurons specific calcium dysregulation. Mol Cell Ther. 2014. 2: 26-
133. Johnson FO, Atchison WD. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology. 2009. 30: 761-5
134. Johnson WG. The clinical spectrum of hexosaminidase deficiency diseases. Neurology. 1981. 31: 1453-6
135. Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011. 7: e1002214-
136. Kadoyama K, Funakoshi H, Ohya W, Nakamura T. Hepatocyte growth factor (HGF) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci Res. 2007. 59: 446-56
137. Kaji R, Kodama M, Imamura A, Hashida T, Kohara N, Ishizu M. Effect of ultrahigh-dose methylcobalamin on compound muscle action potentials in amyotrophic lateral sclerosis: A double-blind controlled study. Muscle Nerve. 1998. 21: 1775-8
138. Kamel F, Umbach DM, Munsat TL, Shefner JM, Hu H, Sandler DP. Lead exposure and amyotrophic lateral sclerosis. Epidemiology. 2002. 13: 311-9
139. Kang JD, Bohlman HH. Cervical spondylotic myelopathy. Curr Opin Orthop. 1996. 7: 13-21
140. Kasarskis EJ, Scarlata D, Hill R, Fuller C, Stambler N, Cedarbaum JM. A retrospective study of percutaneous endoscopic gastrostomy in ALS patients during the BDNF and CNTF trials. J Neurol Sci. 1999. 169: 118-25
141. Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010. 131: 517-26
142. Kiaei M, Kipiani K, Calingasan NY, Wille E, Chen J, Heissig B. Matrix metalloproteinase-9 regulates TNF-alpha and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2007. 205: 74-81
143. Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis. 2005. 2: 246-54
144. Kiaei M. Peroxisome proliferator-activated receptor-gamma in amyotrophic lateral sclerosis and Huntington's disease. PPAR Res 2008. 2008. p.
145. Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005. 169: 561-7
146. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O. Amyotrophic lateral sclerosis. Lancet. 2011. 377: 942-55
147. Kirkwood SC, Su JL, Conneally P, Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol. 2001. 58: 273-8
148. Kirschner A.editors. Spinal Bulbar Muscular Atrophy: Kennedy Disease, Genetic Counseling for Adult Neurogenetic Disease. New York Heiderlberg Dorddrecht. London: Springer; 2015. p. 183-93
149. Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999. 5: 347-50
150. Kobayashi M, Ikeda K, Kinoshita M, Iwasaki Y. Amyotrophic lateral sclerosis with supranuclear ophthalmoplegia and rigidity. Neurol Res. 1999. 21: 661-4
151. Körner S, Kollewe K, Ilsemann J, MüllerHeine A, Dengler R, Krampfl K. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur J Neurol. 2013. 20: 647-54
152. Krakora D, Mulcrone P, Meyer M, Lewis C, Bernau K, Gowing G. Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol Ther. 2013. 21: 1602-10
153. Kübler A, Winter S, Ludolph AC, Hautzinger M, Birbaumer N. Severity of depressive symptoms and quality of life in patients with amyotrophic lateral sclerosis. Neurorehabil Neural Repair. 2005. 19: 182-93
154. Kühnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008. 4: 366-74
155. Kurlander HM, Patten BM. Metals in spinal cord tissue of patients dying of motor neuron disease. Ann Neurol. 1979. 6: 21-4
156. Kurt A, Nijboer F, Matuz T, Kübler A. Depression and anxiety in individuals with amyotrophic lateral sclerosis: Epidemiology and management. CNS Drugs. 2007. 21: 279-91
157. Kurtzke JF. Epidemiology of amyotrophic lateral sclerosis. Adv Neurol. 1982. 36: 281-302
158. Kuzuhara S, Kokubo Y, Sasaki R, Narita Y, Yabana T, Hasegawa M. Familial amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Kii Peninsula of Japan: Clinical and neuropathological study and tau analysis. Ann Neurol. 2001. 49: 501-11
159. Kuzuhara S, Kokubo Y. Atypical parkinsonism of Japan: Amyotrophic lateral clerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): An update. Mov Disord. 2005. 20: S108-13
160. Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009. 323: 1205-8
161. Lacomblez L, Bensimon G, Douillet P, Doppler V, Salachas F, Meininger V. Xaliproden in amyotrophic lateral sclerosis: Early clinical trials. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004. 5: 99-106
162. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996. 347: 1425-31
163. Lauria G, Campanella A, Filippini G, Martini A, Penza P, Maggi L. Erythropoietin in amyotrophic lateral sclerosis: A pilot, randomized, double-blind, placebo-controlled study of safety and tolerability. Amyotroph Lateral Scler. 2009. 10: 410-5
164. Leigh PN, Abrahams S, Al-Chalabi A, Ampong MA, Goldstein LH, Johnson J. The management of motor neurone disease. J Neurol Neurosurg Psychiatry. 2003. 74: iv32-iv47
165. Leigh PN, Ray-Chaudhuri K. Motor neuron disease. J Neurol Neurosurg Psychiatry. 1994. 57: 886-96
166. Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000. 288: 335-9
167. Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998. 20: 589-602
168. Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004. 43: 5-17
169. Logroscino G, Traynor BJ, Hardiman O, Chio’ A, Couratier P, Mitchell JD. Descriptive epidemiology of amyotrophic lateral sclerosis: New evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008. 79: 6-11
170. Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010. 81: 385-90
171. Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002. 59: 1077-9
172. Lorenzl S, Narr S, Angele B, Krell HW, Gregorio J, Kiaei M. The matrix metalloproteinases inhibitor Ro 28-2653 [correction of Ro 26-2853] extends survival in transgenic ALS mice. Exp Neurol. 2006. 200: 166-71
173. Lou JS, Reeves A, Benice T, Sexton G. Fatigue and depression are associated with poor quality of life in ALS. Neurology. 2003. 60: 122-3
174. Louwerse ES, Weverling GJ, Bossuyt PM, Meyjes FE, de Jong JM. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol. 1995. 52: 559-64
175. Ludolph AC, Hugon J, Dwivedi MP, Schaumburg HH, Spencer PS. Studies on the aetiology and pathogenesis of motor neuron diseases 1. Lathyrism: Clinical findings in established cases. Brain. 1987. 110: 149-65
176. Lyall RA, Donaldson N, Polkey MI, Leigh PN, Moxham J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain. 2001. 124: 2000-13
177. Magnus T, Beck M, Giess R, Puls I, Naumann M, Toyka KV. Disease progression in amyotrophic lateral sclerosis: Predictors of survival. Muscle Nerve. 2002. 25: 709-14
178. Magrané J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009. 11: 1615-26
179. Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012. 11: 323-30
180. Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005. 5: 77-87
181. Martin LJ. Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010. 13: 568-80
182. Martínez-Sámano J, Torres-Durán PV, Juárez-Oropeza MA, Verdugo-Díaz L. Effect of acute extremely low frequency electromagnetic field exposure on the antioxidant status and lipid levels in rat brain. Arch Med Res. 2012. 43: 183-9
183. Mathus-Vliegen LM, Louwerse LS, Merkus MP, Tytgat GN, Vianney de Jong JM. Percutaneous endoscopic gastrostomy in patients with amyotrophic lateral sclerosis and impaired pulmonary function. Gastrointest Endosc. 1994. 40: 463-9
184. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000. 1: 120-9
185. Mattson MP. Excitation BolsTORs motor neurons in ALS mice. Neuron. 2013. 80: 1-3
186. Mazzini L, Balzarini C, Colombo R, Mora G, Pastore I, De Ambrogio R. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: Preliminary results. J Neurol Sci. 2001. 191: 139-44
187. Mazzini L, Corrà T, Zaccala M, Mora G, Del Piano M, Galante M. Percutaneous endoscopic gastrostomy and enteral nutrition in amyotrophic lateral sclerosis. J Neurol. 1995. 242: 695-8
188. McDermott C, White K, Bushby K, Shaw P. Hereditary spastic paraparesis: A review of new developments. J Neurol Neurosurg Psychiatry. 2000. 69: 150-60
189. McDonald ER, Wiedenfeld SA, Hillel A, Carpenter CL, Walter RA. Survival in amyotrophic lateral sclerosis. The role of psychological factors. Arch Neurol. 1994. 51: 17-23
190. McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD. Recommended diagnostic criteria for multiple sclerosis: Guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001. 50: 121-7
191. McGuire V, Longstreth WT, Nelson LM, Koepsell TD, Checkoway H, Morgan MS. Occupational exposures and amyotrophic lateral sclerosis. A population-based case-control study. Am J Epidemiol. 1997. 145: 1076-88
192. Meininger V, Asselain B, Guillet P, Leigh PN, Ludolph A, Lacomblez L. Pentoxifylline in ALS: A double-blind, randomized, multicenter, placebo-controlled trial. Neurology. 2006. 66: 88-92
193. Meininger V, Drory VE, Leigh PN, Ludolph A, Robberecht W, Silani V. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: A double- blind, randomized, multicentre, placebo-controlled trial. Amyotroph Lateral Scler. 2009. 10: 378-83
194. Mellies U, Ragette R, Schwake C, Baethmann M, Voit T, Teschler H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology. 2001. 57: 1290-5
195. Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015. 14: 478-84
196. Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002. 125: 1522-33
197. Michal Freedman D, Kuncl RW, Weinstein SJ, Malila N, Virtamo J, Albanes D. Vitamin E serum levels and controlled supplementation and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013. 14: 246-51
198. Millan MJ. The induction of pain: An integrative review. Prog Neurobiol. 1999. p. 1-164
199. Miller R, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database of Systematic Reviews. 2012. 3: CD001447-
200. Miller RG, Bradley W, Cudkowicz M, Meininger V, Mitsumoto H, Sauer D. Phase II/III controlled trial of TCH 346 in patients with amyotrophic lateral sclerosis. Neurology. 2007. 69: 776-84
201. Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009. 73: 1218-26
202. Miller RG, Petajan JH, Bryan WW, Armon C, Barohn RJ, Goodpasture JC. A placebo-controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis.rhCNTF ALS Study Group. Ann Neurol. 1996. 39: 256-60
203. Mills KR. Detecting fasciculations in amyotrophic lateral sclerosis: Duration of observation required. J Neurol Neurosurg Psychiatry. 2011. 82: 549-51
204. Millul A, Beghi E, Logroscino G, Micheli A, Vitelli E, Zardi A. Survival of patients with amyotrophic lateral sclerosis in a population-based registry. Neuroepidemiology. 2005. 25: 114-9
205. Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007. 369: 2031-41
206. Mitchell JD. Amyotrophic lateral sclerosis: Toxins and environment. Amyotroph Lateral Scler. 2000. 1: 235-50
207. Mitsumoto H, Factor-Litvak P, Andrews H, Goetz RR, Andrews L, Rabkin JG. ALS Multicenter Cohort Study of Oxidative Stress (ALS COSMOS): Study methodology, recruitment, and baseline demographic and disease characteristics. Amyotroph Lateral Scler Frontotemporal Degener. 2014. 15: 192-203
208. Mitsumoto H. Diagnosis and progression of ALS. Neurology. 1997. 48: 2S-8S
209. Molina JA, de Bustos F, Jiménez-Jiménez FJ, Gómez-Escalonilla C, García-Redondo A, Esteban J. Serum levels of coenzyme Q10 in patients with amyotrophic lateral sclerosis. J Neural Transm. 2000. 107: 1021-6
210. Mórotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CC. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet. 2012. 21: 1979-88
211. Morozova N, Weisskopf MG, McCullough ML, Munger KL, Calle EE, Thun MJ. Diet and amyotrophic lateral sclerosis. Epidemiology. 2008. 19: 324-37
212. Moser HW, Moser AB, Naidu S, Bergin A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci. 1991. 13: 254-61
213. Last accessed on 2015 Mar 24. Available from: http://www.ninds.nih.gov .
214. Namba T, Nolte CT, Jackrel J, Grob D. Poisoning due to organophosphate insecticides. Acute and chronic manifestations. Am J Med. 1971. 50: 475-92
215. Nefussy B, Artamonov I, Deutsch V, Naparstek E, Nagler A, Drory VE. Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: A pilot study. Amyotroph Lateral Scler. 2009. 15: 1-7
216. Nelson LM, Matkin C, Longstreth WT, McGuire V. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. II. Diet. Am J Epidemiol. 2000. 151: 164-73
217. Newrick PG, Langton-Hewer R. Pain in motor neuron disease. J Neurol Neurosurg Psychiatry. 1985. 48: 838-40
218. Nobile-Orazio E. Multifocal motor neuropathy. J Neuroimmunol. 2001. 115: 4-18
219. Norris FH, Sachais B, Carey M. Trial of baclofen in amyotrophic lateral sclerosis. Archives of Neurology. 1979. 36: 715-6
220. Ochs G, Penn RD, York M, Giess R, Beck M, Tonn J. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000. 1: 201-6
221. Offen D, Barhum Y, Melamed E, Embacher N, Schindler C, Ransmayr G. Spinal cord mRNA profile in patients with ALS: Comparison with transgenic mice expressing the human SOD-1 mutant. J Mol Neurosci. 2009. 38: 85-93
222. Okuda B, Yamamoto T, Yamasaki M, Maya K, Imai T. Motor neuron disease with slow eye movements and vertical gaze palsy. Acta Neurol Scand. 1992. 85: 71-6
223. Oliver D. The quality of care and symptom control – The effects on the terminal phase of ALS/MND. J Neurol Sci. 1996. 139: 134-6
224. Orrell RW, Lane JM, Ross MA. Antioxidant treatment for amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database of Systematic Reviews. 2007. 1: CD002829-
225. Orrell RW, Lane RJ, Ross M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph Lateral Scler. 2008. 9: 195-211
226. Orrell RW. AEOL-10150 (Aeolus). Curr Opin Investig Drugs. 2006. 7: 70-80
227. Orrell RW. Motor neuron disease: Systematic reviews of treatment for ALS and SMA. Br Med Bull. 2010. 93: 145-59
228. O’Toole O, Traynor BJ, Brennan P, Sheehan C, Frost E, Corr B. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry. 2008. 79: 30-2
229. Palomo GM, Manfredi G. Exploring new pathways of neurodegeneration in ALS: The role of mitochondria quality control. Brain Res. 2015. 1607: 36-46
230. Panjabi MM. A hypothesis of chronic back pain: Ligament subfailure injuries lead to muscle control dysfunction. Eur Spine J hypothesis of chronic back pain. 2006. p. 668-76
231. Parboosingh JS, Figlewicz DA, Krizus A, Meininger V, Azad NA, Newman DS. Spinobulbar muscular atrophy can mimic ALS: The importance of genetic testing in male patients with atypical ALS. Neurology. 1997. 49: 568-72
232. Pascuzzi RM, Shefner J, Chappell AS, Bjerke JS, Tamura R, Chaudhry V. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010. 11: 266-71
233. Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004. 43: 19-30
234. Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Rev Neurosci. 2006. 7: 710-23
235. Pfeffer G, Povitz M, Gibson GJ, Chinnery PF. Diagnosis of muscle diseases presenting with early respiratory failure. J Neurol. 2015. 262: 1101-14
236. Phillips JL, Ivaschuk O, Ishida-Jones T, Jones RA, Campbell-Beachler M, Haggren W. DNA damage in Molt-4 Tlymphoblastoid cells exposed to cellular telephone radiofrequency fields in vitro . Bioelectrochem Bioenerg. 1998. 45: 103-10
237. Piepers S, Veldink JH, de Jong SW, van der Tweel I, van der Pol WL, Uijtendaal EV. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann Neurol. 2009. 66: 227-34
238. Polkey MI, Lyall RA, Moxham J, Leigh PN. Respiratory aspects of neurological disease. J Neurol Neurosurg Psychiatry. 1999. 66: 5-15
239. Polymenidou M, Lagier-Tourenne C, Hutt KR, Bennett CF, Cleveland DW, Yeo GW. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012. 1462: 3-15
240. Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: What is it and where are we?. J Clin Invest. 2003. 111: 3-10
241. Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: A mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002. 11: 499-505
242. Rabkin JG, Albert SM, Del Bene ML, O'sullivan I, Tider T, Rowland LP. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005. 65: 62-7
243. Radunovic A, Mitsumoto H, Leigh PN. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. 2007. 6: 913-25
244. Ramirez C, Piemonte ME, Callegaro D, Da Silva HC. Fatigue in amyotrophic lateral sclerosis: Frequency and associated factors. Amyotroph Lateral Scler. 2008. 9: 75-80
245. Reis-Filho J. Next-generation sequencing. Breast Cancer Res. 2009. 11: S12-
246. Riolo SA, Nguyen TA, Greden JF, King CA. Prevalence of depression by race/ethnicity: Findings from the National Health and Nutrition Examination Survey III. Am J Public Health. 2005. 95: 998-1000
247. Riviere M, Meininger V, Zeisser P, Munsat T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol. 1998. 55: 526-8
248. Rizzo F, Riboldi G, Salani S, Nizzardo M, Simone C, Corti S. Cellular therapy to target neuroinflammation in amyotrophic lateral sclerosis. Cell Mol Life Sci. 2014. 71: 999-1015
249. Rollins YD, Oskarsson B, Ringel SP.editors. Primary lateral sclerosis. Blackwell Publishing Ltd; 2009. p. 203-
250. Rosenow EC, Engel AG. Acid maltase deficiency in adults presenting as respiratory failure. Am J Med. 1978. 64: 485-91
251. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001. 344: 1688-700
252. Rowland LP. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1998. 160: S6-24
253. Ryberg H, Askmark H, Persson LI. A double-blind randomized clinical trial in amyotrophic lateral sclerosis using lamotrigine: Effects on CSF glutamate, aspartate, branched-chain amino acid levels and clinical parameters. Acta Neurol Scand. 2003. 108: 1-8
254. Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis?. Brain. 2013. 136: 2342-58
255. Saeed M, Yang Y, Deng HX, Hung WY, Siddique N, Dellefave L. Age and founder effect of SOD1 A4V mutation causing ALS. Neurology. 2009. 72: 1634-9
256. Said G. Chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. 2006. 16: 293-303
257. Salajegheh M, Bryan WW, Dalakas MC. The challenge of diagnosing ALS in patients with prior poliomyelitis. Neurology. 2006. 67: 1078-9
258. Sanjak M, Konopacki R, Capasso R, Roelke KA, Peper SM, Houdek AM. Dissociation between mechanical and myoelectrical manifestation of muscle fatigue in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004. 5: 26-32
259. Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007. 66: 10-6
260. Sau D, De Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, Porrini M. Mutation of SOD1 in ALS: A gain of a loss of function. Hum Mol Genet. 2007. 16: 1604-18
261. Savolainen KM, Loikkanen J, Eerikäinen S, Naarala J. Interactions of excitatory neurotransmitters and xenobiotics in excitotoxicity and oxidative stress: Glutamate and lead. Toxicol Lett. 1998. 102-103: 363-7
262. Schrooten M, Smetcoren C, Robberecht W, Van Damme P. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: A prospective study. Ann Neurol. 2011. 70: 79-83
263. Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol. 2000. 247: I17-27
264. Sheng ZH, Cai Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012. 13: 77-93
265. Shi P, Ström AL, Gal J, Zhu H. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim Biophys Acta. 2010. 1802: 707-16
266. Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Rev. 2004. 45: 250-65
267. Shin JH, Lee JK, Alvaro Estévez.editors. Multiple Routes of Motor Neuron Degeneration in ALS. Current Advances in Amyotrophic Lateral Sclerosis. InTech; 2013. p.
268. Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007. 101: 87-98
269. Simkó M, Mattsson MO. Extremely low frequency electromagnetic fields as effectors of cellular responses in vitro: Possible immune cell activation. J Cell Biochem. 2004. 93: 83-92
270. Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005. 11: 257-70
271. Simons TJ. Cellular interactions between lead and calcium. Br Med Bull. 1986. 42: 431-4
272. Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology. 2004. 62: 1758-65
273. Simpson EP, Yen AA, Appel SH. Oxidative Stress: A common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr Opin Rheumatol. 2003. 15: 730-6
274. Singer MA, Statland JM, Wolfe GI, Barohn RJ. Primary lateral sclerosis. Muscle Nerve. 2007. 35: 291-302
275. Singerman J, Lee L. Consistency of the Babinski reflex and its variants. Eur J Neurol. 2008. 15: 960-4
276. Smith QR, Momma S, Aoyagi M, Rapoport SI. Kinetics of neutral amino acid transport across the blood-brain barrier. J Neurochem. 1987. 49: 1651-8
277. Smith RG, Henry YK, Mattson MP, Appel SH. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 1998. 44: 696-9
278. Sorenson EJ, Windbank AJ, Mandrekar JN, Bamlet WR, Appel SH, Armon C. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008. 71: 1770-5
279. Spencer JP, Jenner A, Butler J, Aruoma OI, Dexter DT, Jenner P. Evaluation of the pro-oxidant and antioxidant actions of L-DOPA and dopamine in vitro: Implications for Parkinson's disease. Free Radic Res. 1996. 24: 95-105
280. Steele AD, Yi CH. Neuromuscular denervation: Bax up against the wall in amyotrophic lateral sclerosis. J Neurosci. 2006. 26: 12849-51
281. Stefanutti D, Benoist MR, Scheinmann P, Chaussain M, Fitting JW. Usefulness of sniff nasal pressure in patients with neuromuscular or skeletal disorders. Am J Respir Crit Care Med. 2000. 162: 1507-11
282. Stommel EW, Cohen JA, Fadul CE, Cogbill CH, Graber DJ, Kingman L. Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: A phase II open label clinical trial. Amyotroph Lateral Scler. 2009. 10: 393-404
283. Sundaram RS, Gowtham L, Nayak BS. The role of excitatory neurotransmitter glutamate in brain physiology and pathology. Asian J Pharm Clin Res. 2012. 5: 1-7
284. Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One. 2007. 2: e689-
285. Talbot K. Motor neuron disease: The bare essentials. Pract Neurol. 2009. 9: 303-9
286. Talbot K. Motor neurone disease. Postgrad Med J. 2002. 78: 513-9
287. Tedman BM, Young CA, Williams IR. Assessment of depression in patients with motor neuron disease and other neurologically disabling illness. J Neurol Sci. 1997. 152: S75-9
288. Thaisetthawatkul P, Logigian EL, Herrmann DN. Dispersion of the distal compound muscle action potential as a diagnostic criterion for chronic inflammatory demyelinating polyneuropathy. Neurology. 2002. 59: 1526-32
289. Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna-Yasek D. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet. 2011. 156B: 285-90
290. Tokuda E, Ono S, Ishige K, Watanabe S, Okawa E, Ito Y. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp Neurol. 2008. 213: 122-8
291. Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci. 2003. 23: 180-92
292. Tovar-Y-Romo LB, Zepeda A, Tapia R. Vascular endothelial growth factor prevents paralysis and motoneuron death in a rat model of excitotoxic spinal cord neurodegeneration. J Neuropathol Exp Neurol. 2007. 66: 913-22
293. Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: A population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003. 74: 1258-61
294. Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC. Neuroprotective agents for clinical trials in ALS: A systematic assessment. Neurology. 2006. 67: 20-7
295. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: A population-based study. Arch Neurol. 2000. 57: 109-13
296. Trias E, Díaz-Amarilla P, Olivera-Bravo S, Isasi E, Drechsel DA, Lopez N. Phenotypic transition of microglia into astrocyte-like cells associated with disease onset in a model of inherited ALS. Front Cell Neurosci. 2013. 7: 274-
297. Trotti D, Rolfs A, Danbolt NC, Brown RH, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999. 2: 427-33
298. Turner MR, Wotton C, Talbot K, Goldacre MJ. Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: Indirect evidence from record linkage study. J Neurol Neurosurg Psychiatry. 2012. 83: 395-8
299. Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008. 70: 144-52
300. Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology. 2005. 65: 1264-7
301. Van den Berg-Vos RM, Visser J, Kalmijn S, Fischer K, de Visser M, de Jong V. A long-term prospective study of the natural course of sporadic adult-onset lower motor neuron syndromes. Arch Neurol. 2009. 66: 751-7
302. Van Den Bosch L, Schwaller B, Vleminckx V, Meijers B, Stork S, Ruehlicke T. Protective effect of parvalbumin on excitotoxic motor neuron death. Exp Neurol. 2002. 174: 150-61
303. van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009. 41: 1083-7
304. Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008. 105: 4022-7
305. Vázquez MC, Ketzoián C, Legnani C, Rega I, Sánchez N, Perna A. Incidence and prevalence of amyotrophic lateral sclerosis in Uruguay: A population-based study. Neuroepidemiology. 2008. 30: 105-11
306. Veldink JH, Kalmijn S, Groeneveld GJ, Wunderink W, Koster A, de Vries JH. Intake of polyunsaturated fatty acids and Vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007. 78: 367-71
307. Vielhaber S, Winkler K, Kirches E, Kunz D, Büchner M, Feistner H. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci. 1999. 169: 133-9
308. Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005. 25: 2463-70
309. Vincent A, Bowen J, Newsom-Davis J, McConville J. Seronegative generalised myasthenia gravis: Clinical features, antibodies, and their targets. Lancet Neurol. 2003. 2: 99-106
310. Visser J, van den Berg-Vos RM, Franssen H, van den Berg LH, Wokke JH, de Jong JM. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007. 64: 522-8
311. von Campenhausen S, Bornschein B, Wick R, Bötzel K, Sampaio C, Poewe W. Prevalence and incidence of Parkinson's disease in Europe. Eur Neuropsychopharmacol. 2005. 15: 473-90
312. Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007. 78: 849-52
313. Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014. 37: 433-42
314. Waibel S, Reuter A, Malessa S, Blaugrund E, Ludolph AC. Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J Neurol. 2004. 251: 1080-4
315. Wang H, O’Reilly ÉJ, Weisskopf MG, Logroscino G, McCullough ML, Schatzkin A. Vitamin E intake and risk of amyotrophic lateral sclerosis: A pooled analysis of data from 5 prospective cohort studies. Am J Epidemiol. 2011. 173: 595-602
316. Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neuroscience. 2004. 125: 191-201
317. Wang W, Zhang F, Li L, Tang F, Siedlak SL, Fujioka H. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J Biol Chem. 2015. 290: 168-82
318. Weisskopf MG, Morozova N, O’Reilly EJ, McCullough ML, Calle EE, Thun MJ. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009. 80: 558-61
319. Weisskopf MG.editors. Formaldehyde exposure and amyotrophic lateral sclerosis. Enironmental and Molecular Mutagensis. NJ USA: Wiley-Blackwell; 2014. 55: S25-
320. Welty DF, Schielke GP, Rothstein JD. Potential treatment of amyotrophic lateral sclerosis with gabapentin: A hypothesis. Ann Pharmacother. 1995. 29: 1164-7
321. Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol. 2004. 3: 93-103
322. Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN, Goldstein LH. Prevalence of depression in a 12-month consecutive sample of patients with ALS. Eur J Neurol. 2007. 14: 993-1001
323. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009. 4: 3-
324. Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999. 2: 50-6
325. Last accessed on 2015 Mar 24. Available from: http://www.who.int/gho/countries/deu/country_profiles/en/ .
326. Worms PM. The epidemiology of motor neuron diseases: A review of recent studies. J Neurol Sci. 2001. 191: 3-9
327. Yang J, Wang Q, Lin L, Wang D, Zheng H, Guan Y. Comparison of clinical and physiological characteristics between Kennedy disease and amyotrophic lateral sclerosis. Nan Fang Yi Ke Da Xue Xue Bao. 2014. 34: 1688-92
328. Yoshino H, Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler. 2006. 7: 241-5
329. Yu Y, Hayashi S, Cai X, Fang C, Shi W, Tsutsui H. Pu-erh tea extract induces the degradation of FET family proteins involved in the pathogenesis of amyotrophic lateral sclerosis. Biomed Res Int 2014. 2014. p.
330. Zhou H, Chen G, Chen C, Yu Y, Xu Z. Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: A meta-analysis. PLoS One. 2012. 7: e48354-
331. Zhu YB, Sheng ZH. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J Biol Chem. 2011. 286: 23432-40
332. Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Samarelli V. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: A population based study. J Neurol Neurosurg Psychiatry. 2008. 79: 33-7
333. Zoccolella S, Santamato A, Lamberti P. Current and emerging treatments for amyotrophic lateral sclerosis. Neuropsychiatr Dis Treat. 2009. 5: 577-95


