

- Department of Clinical Medicine, Nam Dinh University of Nursing, Nam Dinh, Vietnam.
- Department of Medical Biology and Genetics, Hanoi Medical University, Hanoi, Vietnam.
- Department of Neurology, Hanoi Medical University, Hanoi, Vietnam.
- Institute of Theoretical and Applied Research (ITAR), School of Medicine and Pharmacy, Duy Tan University, Da Nang, Vietnam.
Correspondence Address:
Linh Hai Nguyen, Department of Neurology, Hanoi Medical University, Hanoi, Vietnam.
DOI:10.25259/SNI_803_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Anh Tuan Truong1, Anh Thi Lan Luong2, Linh Hai Nguyen3, Huong Van Nguyen3, Diep Ngoc Nguyen4, Ngoc Thi Minh Nguyen2. A novel single-point mutation of NEFH and biallelic SACS mutation presenting as intermediate form Charcot-Marie-Tooth: A case report in Vietnam. 25-Nov-2022;13:553
How to cite this URL: Anh Tuan Truong1, Anh Thi Lan Luong2, Linh Hai Nguyen3, Huong Van Nguyen3, Diep Ngoc Nguyen4, Ngoc Thi Minh Nguyen2. A novel single-point mutation of NEFH and biallelic SACS mutation presenting as intermediate form Charcot-Marie-Tooth: A case report in Vietnam. 25-Nov-2022;13:553. Available from: https://surgicalneurologyint.com/surgicalint-articles/12021/
Date of Submission
01-Sep-2022
Date of Acceptance
04-Nov-2022
Date of Web Publication
25-Nov-2022
Abstract
Background: Charcot-Marie-Tooth disease (CMT) is among the most common group of inherited neuromuscular diseases. SACS mutations were demonstrated to cause autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). However, there have been few case reports regarding to NEFH and SACS gene mutation to CMT in Vietnamese patients, and the diagnosis of CMT and ARSACS in the clinical setting still overlapped.
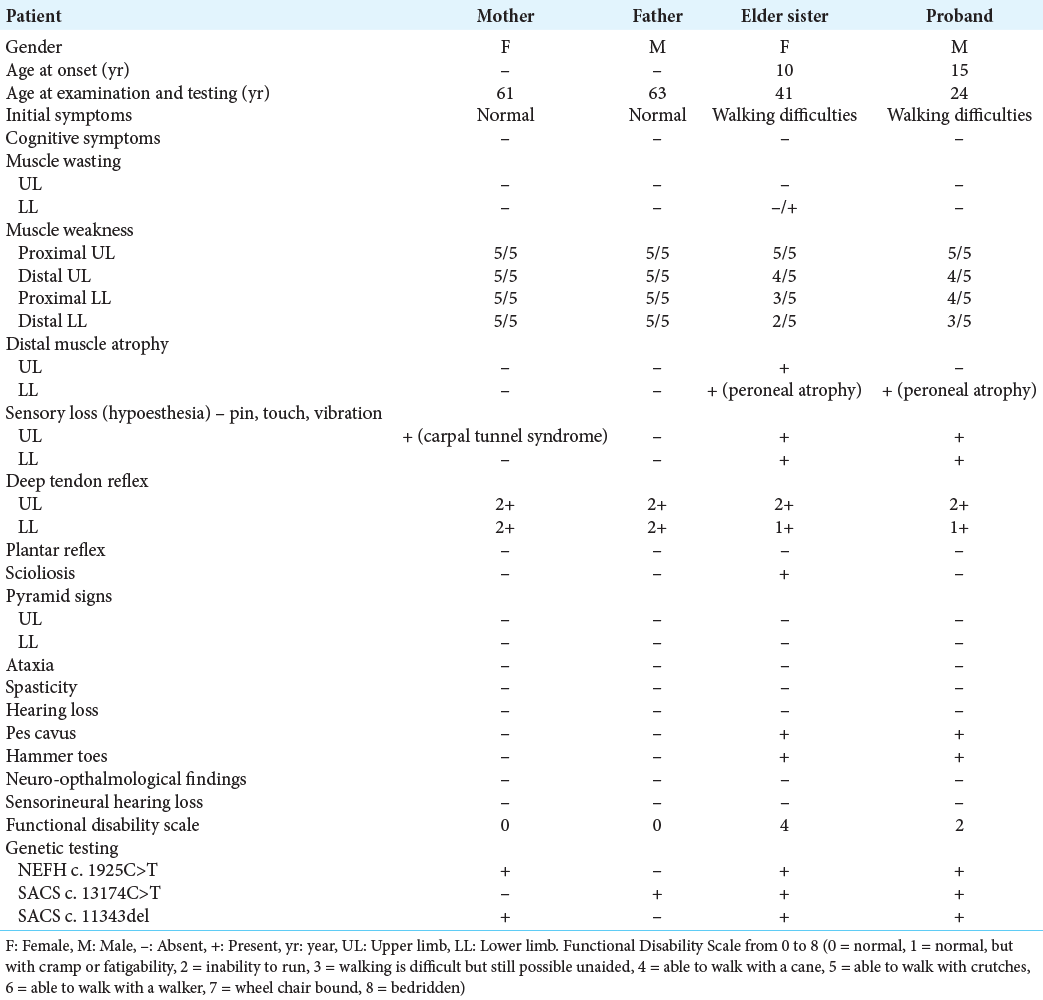
Case Description: We report two patients presenting with sensorimotor neuropathy without cerebellar ataxia, spasticity and other neurological features, being diagnosed with intermediate form CMT by electrophysiological and clinical examination and neuroimaging. By whole-exome sequencing panel of two affected members, and PCR Sanger on NEFH and SACS genes to confirm the presence of selected variants on their parents, we identified a novel missense variant NEFH c.1925C>T (inherited from the mother) in an autosomal dominant heterozygous state, and two recessive SACS variants (SACS c.13174C>T, causing missense variant, and SACS c.11343del, causing frameshift variant) (inherited one from the mother and another from the father) in these two patients. Clinical and electrophysiological findings on these patients did not match classical ARSACS. To the best of our knowledge, this is the first case report of two affected siblings diagnosed with CMT carrying both a novel NEFH variant and biallelic SACS variants.
Conclusion: We concluded that this novel NEFH variant is likely benign, and biallelic SACS mutation (c.13174C>T and c.11343del) is likely pathogenic for intermediate form CMT. This study is also expected to emphasize the current knowledge of intermediate form CMT, ARSACS, and the phenotypic spectrum of NEFH-related and SACS-related disorders. We expect to give a new understanding of CMT; however, further research should be conducted to provide a more thorough knowledge of the pathogenesis of CMT in the future.
Keywords: Autosomal recessive spastic ataxia of Charlevoix-Saguenay, Intermediate Charcot-Marie-Tooth disease, NEFH, Novel variant, SACS
INTRODUCTION
Hereditary motor and sensory neuropathies, classically known as Charcot-Marie-Tooth disease (CMT), are among the most common group of inherited neuromuscular diseases. These disorders are often characterized by progressive distal muscle atrophy and weakness, mild-to-moderate distal sensory loss, and foot deformities.[
Neurofilaments (NF) were the major scaffolding protein, with a diameter of 10 mm, almost exclusively expressed in the neuronal skeleton, classified into heavy (NEFH), medium (NEFM), and light (NEFL) chains based on the molecular weight. The NFs were formed in the neuronal cell body, and transported to the axons, forming the neuronal network structures to maintain the structural, functional, and electrical integrity of axons. Of those, NEFL gene mutation was confirmed to cause CMT1F and CMT2 with impaired axonal transport.[
Furthermore, single nucleotide polymorphisms and mutation variants may explain the structural and functional changes associated with the increased risk of these diseases. Several reports showed the relation between NEFH as a biomarker of neuronal damage related to intermediate form CMT and ALS;[
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is one rare neurodegenerative disorder which belongs to spastic ataxia syndrome. Spastic ataxia is characterized by the combination of cerebellar ataxia with spasticity and other pyramidal features. ARSACS (MIM: 270550) is characterized by early-onset spastic ataxia and sensorimotor neuropathy.[
Sacsin (SACS) gene is located on chromosome 13, comprising 10 exons, encoding a large multidomain protein called sacsin. Sacsin is expressed in different tissues, notably in the central nervous system. In the brain, sacsin is most expressed in the motor system, including the granular cerebellar system and Purkinje cells.[
Recently, the phenotypical spectrum of SACS variants has been expanded thanks to revolutions in genetic testing. Diverse clinical features are not only framed in the classic triad of ARSACS described in the literature (cerebellar ataxia, spasticity, and peripheral neuropathy), atypical features such as deafness, seizures, and mental retardation have been reported.[
In the literature, inherited peripheral neuropathies (in which the most common entity is CMT) were described as the disorder between Schwann cells and axons in peripheral nerves, resulting in abnormalities in myelin assembly and axonal transport, causing peripheral demyelination and peripheral axonopathy.[
Concerning the overlap between ARSACS and CMT in the clinical setting to diagnose a patient presenting with hereditary sensorimotor neuropathy, herein, we report a case of intermediate form CMT regarding a highly pathogenic cause of biallelic SACS mutations and a likely benign novel NEFH variant and address several limitations during diagnosis due to restriction of condition in a low- and middle-income country like Vietnam. Thus, a genetic panel to aid in diagnosing CMT and neurological diseases, particularly in the clinical setting of developing countries, should be optimized.
CASE DESCRIPTION
Two patients (one female and one male) were two nonconsanguineous siblings in one family with normal neuropsychological development, examined by two authors. Their family history, history, and history of exposure were unremarkable. Deceased or alive family members within three generations who had the same symptoms were not reported by the patients. Nerve conduction studies (NCS) and electromyography (EMG) were performed using standard techniques.
Definition
Definite conduction blocks were defined as a decrease of more than 50% in distal compound muscle action potential (CMAP) amplitude. Then, probable conduction blocks were defined as a decrease of 30–50% in distal CMAP amplitude.
DNA samples were extracted from peripheral blood, and whole-exome sequencing (WES) neuropathy multigene panel was performed on two affected members. Due to restriction of finance, we only utilized PCR Sanger sequencing to confirm the presence of preselected variants (NEFH variants and two SACS variants identified in the WES panel of two affected members) on their parents, who were in normal-phenotype.
A database of clinical variant alleles was utilized to search for the variants’ allele frequencies and interpretation.
Written informed consent forms were obtained from the proband’s family for participation in this case report, in agreement with local ethic committees of Hanoi Medical University Hospital, Vietnam, and with the 1964 Helsinki Declaration and its later amendments.
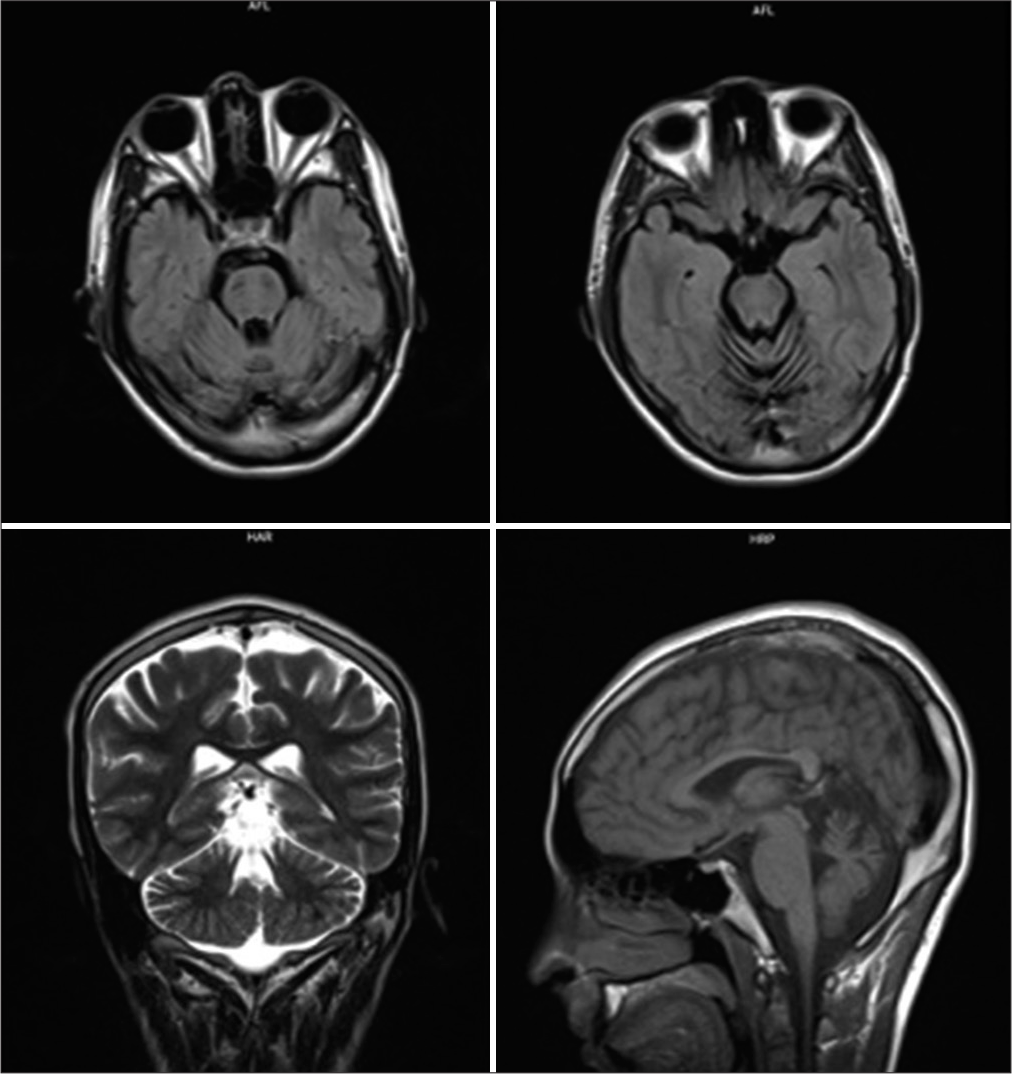
Clinical and neuroimaging findings
The pedigree in this paper is summarized in
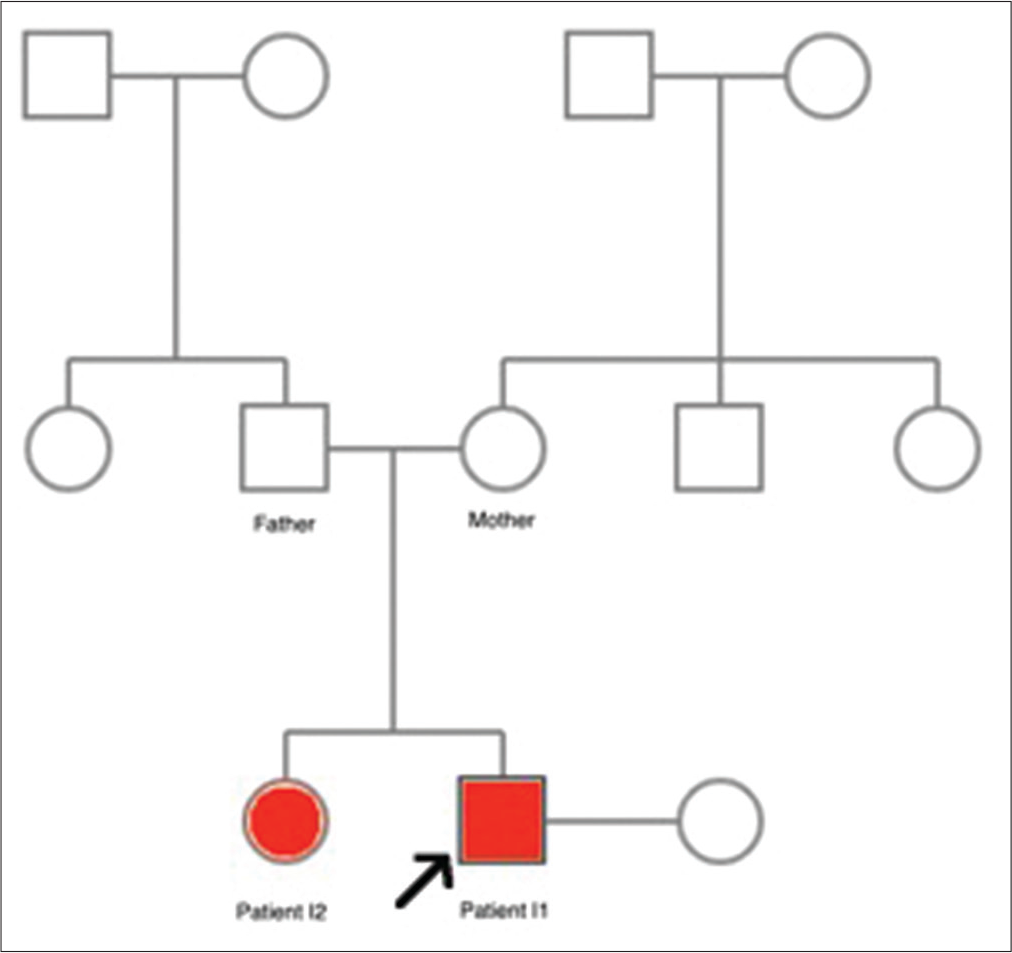
Patient I2
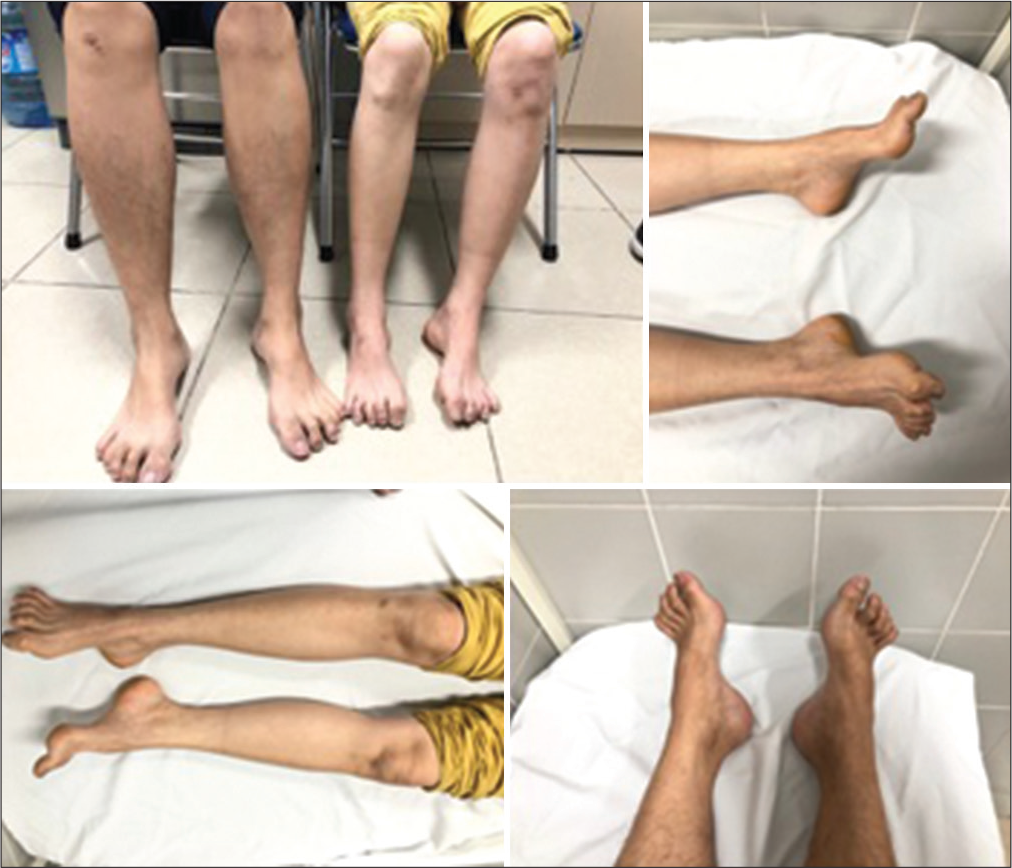
The proband, a 24-year-old male, presented to our hospital with walking difficulties due to LL weakness. He reported no significant symptoms until he was 15; he first recognized slight LL weakness and progressive foot deformities, affecting his daily life and schooling unremarkably, so he did not go to the hospital for examination. At the age of 20, he developed weakness of predominantly distal LLs, weak bilateral high-arched feet, and unable to dorsiflex. At the last examination, he could walk and run slowly without aid. Neurological examination revealed distal muscle atrophy predominantly in the LLs; pes cavus, and hammer toes; distal muscle weakness of four limbs, predominantly LLs (UL: 4/5 at distal and 5/5 at proximal; whereas LL: 3/5 at distal and 4/5 at proximal); distal hypoesthesia in a stocking-and-glove distribution; reduced deep tendon reflexes, no Babinski sign, no spasticity, and no cerebellar and gait ataxia [
Patient I1
Until after the proband’s first presentation to the hospital, we consulted him to bring all of his family members for a comprehensive neurological consultation. The proband’s elder sister, a 41-year-old unmarried female, presented to the hospital with severe weakness in four limbs. She was unsure about the onset and dropped out of schooling at 16 because of financial burden. Neurological examination revealed atrophy of thenar eminence on both hands, pes cavus, hammer toes, Achilles contractures, and distal muscle weakness of four limbs. Laboratory tests remained within normal range. She presented with similar findings but more severe than her brother’s. Their clinical examination images are also shown in [
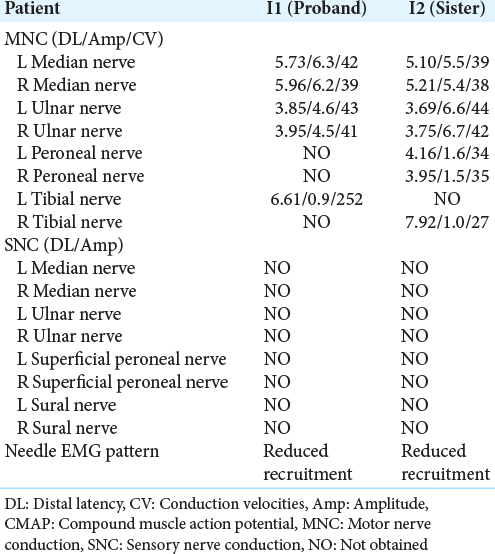
Electrophysiological findings
A standardized electrophysiological study was performed on the entire family, showing unremarkable in both parents. The results of sensory and motor NCS and EMG in the affected members are displayed in [
Sural nerve biopsy
We could not perform nerve biopsy on these affected patients because this invasive diagnostic technique was not authorized in Vietnam. We are in the process of demanding permission to conduct this technique soon at our medical center, and we expect it to happen quickly.
WES
We carried out WES on the DNA of the proband and his elder sister, prioritizing variants of genes known to be responsible for CMT and neuromuscular disorders [
cDNA level: NM_021076.4: c.1925C>T gDNA level: Chr22 (29489565) Protein level: p.Thr642Met
We also identified two heterozygous SACS mutation (a missense mutation SACS c.13174C>T in exon 2, and a frame shift mutation SACS c.11343del in exon 2) from the probands’s and his elder sister’s SACS gene, both of which indicate the autosomal recessive inheritance pattern.
cDNA level: NM_001278055.2: c.13174C>T gDNA level: Chr13 (23330261) Protein level: p.Pro4392Ser
And: cDNA level: NM_001278055.2: c.11343del
gDNA level: Chr13 (23332092) Protein level: p.His3782IlefsTer15
Initially, we did not expect biallelic SACS mutations to be pathogenic due to the absence of clinical and electrophysiological findings supporting the diagnosis of spastic ataxia in these two patients. PCR Sanger on preselected NEFH and SACS variants for the proband’s parents to confirm the presence of these variants, concluding that SACS c.13174C>T variant inherited from the father, and NEFH c.1925C>T and SACS c.11343del inherited from the mother [
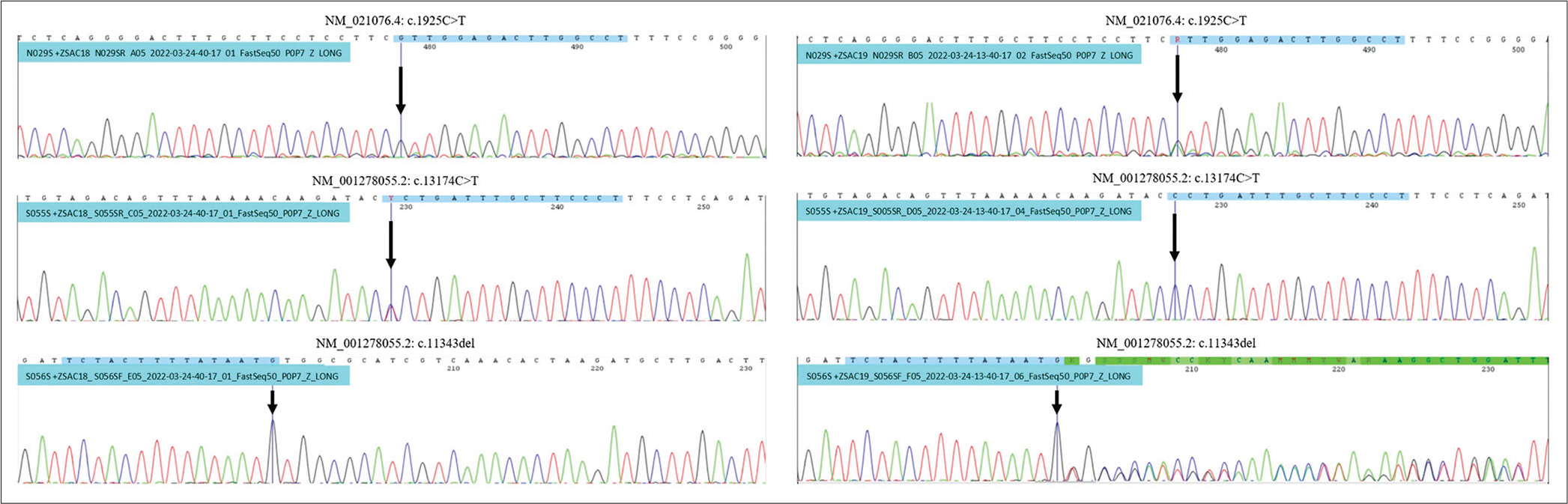
Figure 4:
Analysis of PCR Sanger to confirm the presence of these variants on their parents revealed, both patients were inherited NEFH c.1925C>T variant in autosomal recessive trait, and SACS c.11343del variant from the mother. SACS c.13174C>T variant was inherited from the father. Left PCR belongs to the father, right PCR belongs to the mother.
We noticed that the affected patients carried NEFH variant and biallelic SACS variants while the normal-phenotype persons did not. Based on using bioinformatics variant analysis tools: Mutation Taster
SACS mRA and sacsin quantification
We notice that SACS mRA and sacsin quantification are unavailable in our laboratory.
DISCUSSION
Mutations in NF genes are recently reported to be the likely causative mutant of neurodegenerative diseases, peripheral neuropathies, anterior horn cell diseases, and Parkinson’s disease.[
In this case report, two affected members met the diagnostic criteria of intermediate form CMT, including: (1) regarding clinical features, symmetrical, distal motor and sensory involvement, predominantly in the LLs, absence of ataxia, absence of the upper motor neuron signs, absence of spasticity, and age of onset ranging from 3 to 43 years; (2) regarding electrophysiological findings, needle EMG and NCS consistent with both axonal and demyelinating lesion with distal motor and sensory axonal neuropathy, and moderately reduced MCV ranging from 38 to 42 m/s. These findings were consistent with the previous reports of intermediate form CMT.[
After genetic testing of the family members, we also combine several bioinformatics tools to aid in diagnosis. We found that NEFH c.1925C>T in the proband and his elder sister was inherited from the normal phenotype mother, suggesting that this novel variant was likely benign. SACS variants in the proband and his elder sister were inherited in an autosomal recessive pattern. These affected members possessed biallelic SACS mutation. ClinVar database predicted this novel NEFH c.1925C>T variant was “benign,” and the other SACS variants were “likely pathogenic.” Hence, we safely conclude that the NEFH c.1925C>T variant identified in the proband’s family was considered as “benign to CMT” and autosomal dominant trait, and biallelic SACS mutation was considered as “pathogenic to CMT” and autosomal recessive trait.
Most recent studies on CMT report the insertion or deletion fragments at 3 kb, which caused a frameshift and may affect the 3’UTR region on NEFH gene. One report by Yan et al. showed a single-point NEFH mutation causing CMT, suggesting the hypothesis that nucleotides encoded by NEFH near 2 kb may still alter final protein structures, resulting in neurological disorders.[
In addition, several genetic mutations are shown to be prone to muscular damage, for example, NEFL mutations whose CK activity increases with axonal damage and correlates with muscle tissue changes. It was concluded that increased CK activity was often present with axonal phenotypes.[
Concerning the definitive diagnosis of CMT or ARSACS with pure sensorimotor neuropathy (CMT-like ARSACS), ARSACS classically represented ataxia and spastic paraplegia. However, one report by Souza et al. reported two cases of unrelated patients with CMT carrying homozygous SACS mutation c.11542_11544 del [p.Ile3848del], and questioned that SACS gene mutation may cause early-onset pure axonal neuropathy, with classical neuroimaging findings, but without other neurological features, hence emphasizing the importance of neuroimaging in the definitive diagnosis of ARSACS versus CMT. Few studies reported ARSACS with pure axonal neuropathy and the role of SACS gene mutations in correlation with phenotype. In our case study, the affected patients also carried a novel NEFH variant. We were concerned whether the novel NEFH c.1925C>T may be genetic epistasis with biallelic SACS c.13174C>T and SACS c.11343del to CMT-like ARSACS is still unclear; thus, the further role of each variant should be investigated carefully.
A recent study in 2021 by Longo et al. has demonstrated that sacsin is absent in patients with ARSACS, and SACS mRNA is reduced in fibroblasts carrying variants. The authors proposed that the evaluation of sacsin levels should be included in clinical genetic practice to establish a definite ARSACS diagnosis. However, this quantification technique is still unavailable in our country and remains a challenge for our diagnosis.[
Due to the restriction of financial support for the study, only the proband and his elder sister underwent a WES panel of neuromuscular disorders (which cost 300 USD for each), the parents underwent NEFH c.1925C>T and SACS c.13174C>T and SACS c.11343del site verification by PCR Sanger (which cost at a lower fee of 60 USD for each; however, the Department of Medical Genetics has sponsored these performances). Moreover, only two members in one generation within three generations were affected, and we still have to wait for the clinical phenotype of the proband’s child (because his elder sister decided not to marry and give birth). Therefore, the genetic information was insufficient, and whether the above three variants were family-inherited or distinct cannot be determined. The relevant clinical evidence cannot support the variants’ pathogenicity to intermediate form CMT. We will follow-up with the proband’s family for predicted intermediate form CMT on his child, practice medical genetic consultation on the proband’s couple, and screen for other diseases which may sprout in the future.
For the drawbacks of our study, because the variant is rare, and only two among four family members were reported with CMT, the sample size was too small to implicate the variant pathogenic effect from a genetic point of view. Second, we could not analyze the proband’s parents to discover further variants other than three pointed mutations from the proband and his elder sister. Third, the study did not implement a sural nerve biopsy due to current unavailability and authorization in Vietnam. Forth, the diagnosis of CMT and ARSACS in a clinical setting remains a challenge. Although analysis of several reports on the association of NEFH mutations and intermediate CMT was reviewed above to suggest that biallelic SACS mutation may play a role in the pathogenesis of CMT, it is still challenging to explain the pathogenesis of CMT and related disorders.
CONCLUSION
Our study was the first report in Vietnam associated with intermediate form CMT, which emphasizes the importance of genetic testing and neuroimaging to aid in the diagnosis of CMT versus ARSACS in patients presenting with pure sensorimotor neuropathy, and expects to expand the current understanding about NEFH and SACS-related clinical spectrums. It is also the first report of two patients with CMT presenting with a novel NEFH variant with biallelic SACS mutations, raising concern about genetic epistasis in the pathophysiological mechanism of CMT. Further studies should be conducted to diagnose and reveal the exact pathogenesis of these diseases.
Ethical standards
This study was approved by the Institutional Review Board of Hanoi Medical University and informed consents were obtained from all participating members.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Al-Chalabi A, Miller CC. Neurofilaments and neurological disease. Bioessays. 2003. 25: 346-55
2. Aruta F, Severi D, Iovino A, Spina E, Barghigiani M, Ruggiero L. Proximal weakness involvement in the first Italian case of Charcot-Marie-Tooth 2CC harboring a novel frameshift variant in NEFH. J Peripher Nerv Syst. 2021. 26: 231-4
3. Bian X, Lin P, Li J, Long F, Duan R, Yuan Q. Whole-genome linkage analysis with whole-exome sequencing identifies a novel frameshift variant in NEFH in a Chinese family with Charcot-Marie-Tooth 2: A novel variant in NEFH for Charcot-Marie-Tooth 2. Neurodegener Dis. 2018. 18: 74-83
4. Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. 1978. 5: 61-9
5. Bouhlal Y, Amouri R, Euch-Fayeche GE, Hentati F. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: An overview. Parkinsonism Relat Disord. 2011. 17: 418-22
6. Charalampopoulou A, Veltsista D, Taraviras S, Chroni E. First case report of Charcot-Marie-Tooth disease Type 2CC with a frameshift mutation of NEFH gene in Greece. Neurol Sci. 2021. 42: 4377-9
7. Chen CX, Dong HL, Wei Q, Li LX, Yu H, Li JQ. Genetic spectrum and clinical profiles in a Southeast Chinese cohort of Charcot-Marie-Tooth disease. Clin Genet. 2019. 96: 439-48
8. Elbracht M, Senderek J, Schara U, Nolte K, Klopstock T, Roos A. Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot-Marie-Tooth disease Type 2E. Clin Neuropathol. 2014. 33: 335-43
9. Ericson U, Ansved T, Borg K. Charcot-Marie-Tooth disease--muscle biopsy findings in relation to neurophysiology. Neuromuscul Disord. 1998. 8: 175-81
10. Harding AE, Thomas PK. Genetic aspects of hereditary motor and sensory neuropathy (Types I and II). J Med Genet. 1980. 17: 329-36
11. Hu RT, Yu Q, Zhou SD, Yin YX, Hu RG, Lu HP. Co-expression network analysis reveals novel genes underlying Alzheimer’s disease pathogenesis. Front Aging Neurosci. 2020. 12: 605961
12. Ikenberg E, Reilich P, Abicht A, Heller C, Schoser B, Walter MC. Charcot-Marie-Tooth disease Type 2CC due to a frameshift mutation of the neurofilament heavy polypeptide gene in an Austrian family. Neuromuscul Disord. 2019. 29: 392-7
13. Jacquier A, Delorme C, Belotti E, Juntas-Morales R, Solé G, Dubourg O. Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathol Commun. 2017. 5: 55
14. Kuchay RA, Mir YR, Zeng X, Hassan A, Musarrat J, Parwez I. ARSACS as a worldwide disease: Novel SACS mutations identified in a consanguineous family from the remote tribal Jammu and Kashmir region in India. Cerebellum. 2019. 18: 807-12
15. Larivière R, Gaudet R, Gentil BJ, Girard M, Conte TC, Minotti S. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum Mol Genet. 2015. 24: 727-39
16. Lin F, Lin W, Zhu C, Lin J, Zhu J, Li XY. Sequencing of neurofilament genes identified NEFH Ser787Arg as a novel risk variant of sporadic amyotrophic lateral sclerosis in Chinese subjects. BMC Med Genomics. 2021. 14: 222
17. Longo F, Ritis DD, Miluzio A, Fraticelli D, Baets J, Scarlato M. Assessment of sacsin turnover in patients with ARSACS: Implications for molecular diagnosis and pathogenesis. Neurology. 2021. 97: e2315-27
18. Maier M, Berger P, Suter U. Understanding Schwann cell-neurone interactions: The key to Charcot-Marie-Tooth disease?. J Anat. 2002. 200: 357-66
19. Nam DE, Jung SC, Yoo DH, Choi SS, Seo SY, Kim GH. Axonal Charcot-Marie-Tooth neuropathy concurrent with distal and proximal weakness by translational elongation of the 3’ UTR in NEFH. J Peripher Nerv Syst. 2017. 22: 200-7
20. Nan H, Takaki R, Hata T, Ichinose Y, Tsuchiya M, Koh K. Novel GARS mutation presenting as autosomal dominant intermediate Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2019. 24: 156-60
21. Nicholson G, Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: A review. Neuromolecular Med. 2006. 8: 123-30
22. Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo JM. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet. 2009. 18: 1556-65
23. Pezzini F, Bianchi M, Benfatto S, Griggio F, Doccini S, Carrozzo R. The networks of genes encoding palmitoylated proteins in axonal and synaptic compartments are affected in PPT1 overexpressing neuronal-like cells. Front Mol Neurosci. 2017. 10: 266
24. Pilliod J, Moutton S, Lavie J, Maurat E, Hubert C, Bellance N. New practical definitions for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Ann Neurol. 2015. 78: 871-86
25. Rebelo AP, Abrams AJ, Cottenie E, Horga A, Gonzalez M, Bis DM. Cryptic amyloidogenic elements in the 3’ UTRs of neurofilament genes trigger axonal neuropathy. Am J Hum Genet. 2016. 98: 597-614
26. Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013. 9: 562-71
27. Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schüle R. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis. 2013. 8: 41
28. Vermeer S, Meijer RP, Pijl BJ, Timmermans J, Cruysberg JR, Bos MM. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics. 2008. 9: 207-14
29. Yan J, Qiao L, Peng H, Liu A, Wu J, Huang J. A novel missense pathogenic variant in NEFH causing rare Charcot-Marie-Tooth neuropathy Type 2CC. Neurol Sci. 2021. 42: 757-63

