

- Departments of Neurosurgery, Shinshu University, School of Medicine, Asahi, Matsumoto, Nagano, Japan.
- Departments of Pathology, Shinshu University, School of Medicine, Asahi, Matsumoto, Nagano, Japan.
Correspondence Address:
Toshihiro Ogiwara
Departments of Neurosurgery, Shinshu University, School of Medicine, Asahi, Matsumoto, Nagano, Japan.
DOI:10.25259/SNI_474_2019
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Tobechi Mbadugha, Toshihiro Ogiwara, Alhusain Nagm, Takatoshi Hasegawa, Keisuke Kamiya, Yuki Matsumoto, Mikiko Kobayashi, Kazuhiro Hongo. Acromegaly in preadolescence: A case report of a 9-year-old boy with acromegaly. 04-Apr-2020;11:61
How to cite this URL: Tobechi Mbadugha, Toshihiro Ogiwara, Alhusain Nagm, Takatoshi Hasegawa, Keisuke Kamiya, Yuki Matsumoto, Mikiko Kobayashi, Kazuhiro Hongo. Acromegaly in preadolescence: A case report of a 9-year-old boy with acromegaly. 04-Apr-2020;11:61. Available from: https://surgicalneurologyint.com/surgicalint-articles/9944/
Date of Submission
02-Sep-2019
Date of Acceptance
20-Mar-2020
Date of Web Publication
04-Apr-2020
Abstract
Background: Acromegaly has been reported in adolescents and young adults, but it is unusual in preadolescence. Diagnosis and management pose different challenges in this age group. Here, we present a rare case of acromegaly in preadolescence.
Case Description: A 9-year-old boy presented with acromegalic features and MRI revealed a pituitary tumor. He was diagnosed as having growth hormone secreting pituitary adenoma based on the multidisciplinary assessment, and underwent gross total tumor resection through an endoscopic endonasal approach (EEA) with subsequent normalization of the hormonal parameters.
Conclusions: Advances in EEA have made safe removal and cure possible even in children. However, long- term follow-up is needed in such younger patients with multidisciplinary management by neurosurgeons, endocrinologists, pediatricians, and ENT surgeons.
Keywords: Acromegaly, Endoscopic endonasal approach, Growth hormone secreting pituitary adenoma, Preadolescence
INTRODUCTION
Pituitary adenomas are uncommon in childhood accounting for about 2.5% of supratentorial tumors in children.[
CASE PRESENTATION
History and examination
A 9-year-old boy presented with acromegalic features and rapid height growth discovered 4 months before presentation during school physical examination. He had complained of intermittent headache and had a recent history of snoring during sleep. The parents and siblings had normal height and physical features with no history of endocrinopathies. On examination, he had a height of 151.8 cm (more than 2SD for age), weight of 40.2 kg, and hypertrophy of the lips. The patient had a shoe size of 24.5 cm (corresponding to the size appropriate for a Japanese boy of about 15 years old). The patient’s visual assessment was normal. Examinations of the cardiovascular and other systems yielded essentially normal results.
Laboratory and radiological diagnosis
Hormonal assay showed marked elevation of the levels of growth hormone (GH) and insulin-like growth factor (IGF-1). Random GH was 25.0 ng/ml, IGF-1 was 873 (99–423 ng/ml), with nadir GH during 75-g oral glucose tolerance test (75 g OGTT) of 15.9 ng/ml. Apart from the moderate increase in free T3 (5.27 pg/ml), other pituitary hormones were within the normal limits. Magnetic resonance imaging (MRI) of the brain revealed a pituitary tumor measuring 12 × 10 × 9 mm, without cavernous sinus invasion [
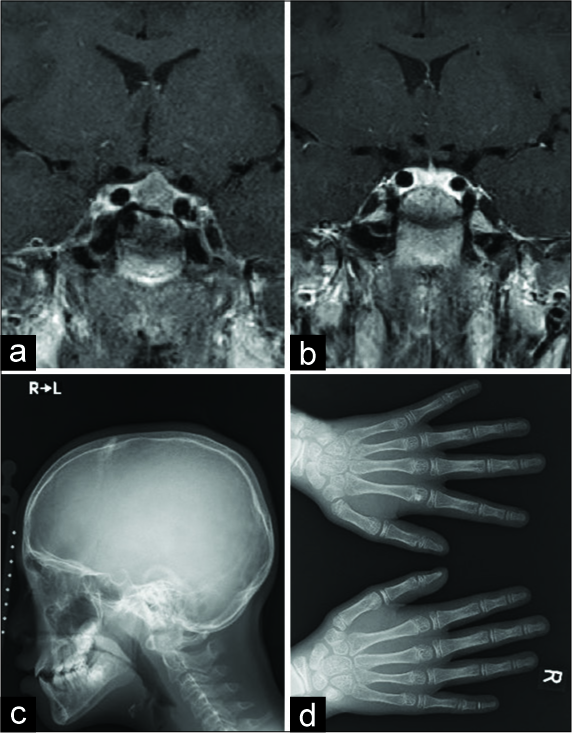
Figure 1:
Preoperative MRI showing a sellar mass lesion with less enhancement with gadolinium (a). Postoperative T1- weighted postcontrast image showing complete tumor removal with preservation of the pituitary gland and stalk (b). Skull X-ray showing prominence of the jaw and mild frontal bossing (c). Hand X-ray revealing cauliflower appearance at the distal phalanx of the hands (d).
Treatment and postoperative course
A preoperative diagnosis of GH-secreting macroadenoma was made, and tumor resection was performed through an endoscopic endonasal approach (EEA). Although the working space during the nasal and sphenoid phase was very narrow due to the preadolescent age and acromegalic features, it was possible to manipulate surgical instruments smoothly during sellar phase. The tumor was whitish and soft, and internal debulking was done using a double-suction technique. The pseudocapsule was also removed, and finally gross total resection of the tumor was performed [
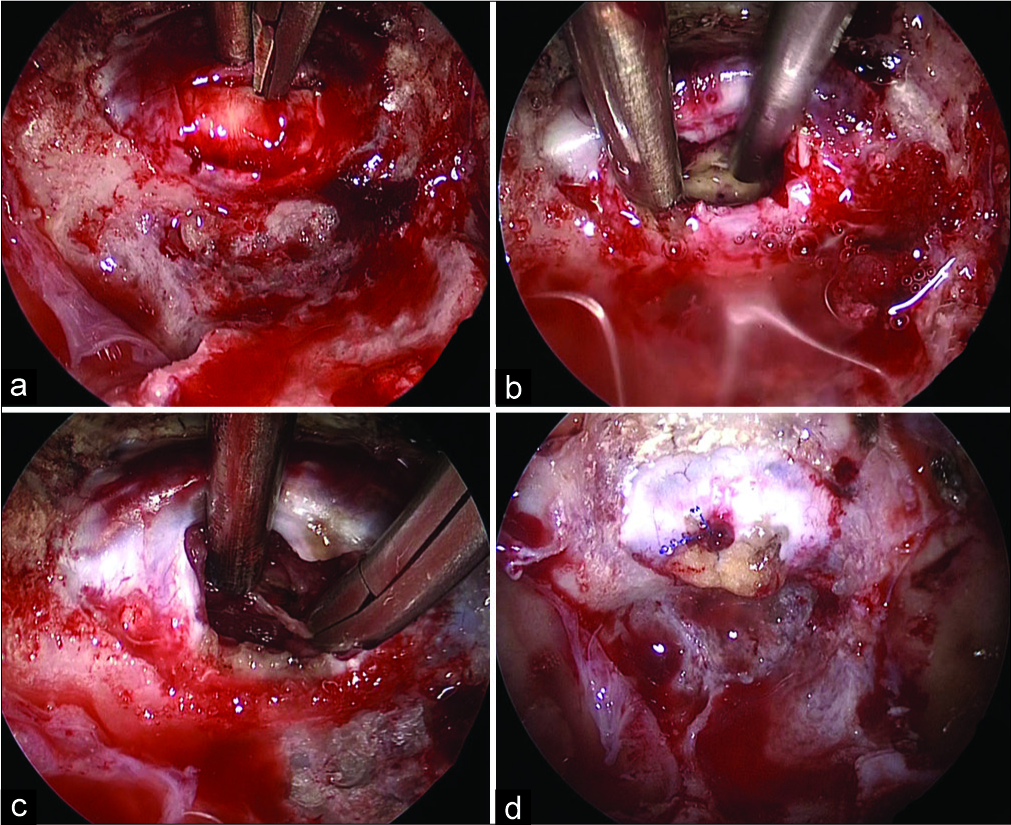
Figure 2:
Intraoperative photographs. The stretched anterior pituitary lobe had covered the tumor (a). The tumor was resected in a double-suction technique intracapsularly (b). The pseudocapsule was resected following internal debulking of the tumor (c). A fat graft was packed into the cavity with a dural stitch following tumor resection (d).

Figure 3:
Histopathological photographs. The tumor was composed of monomorphic neoplastic cells with eosinophilic cytoplasm (H and E, ×100) (a). Most of the neoplastic cells showed immunoreactivity for GH in their cytoplasm (immunohistochemistry, ×100) (b). The Ki-67 proliferation index was 3% (22/725) (immunohistochemistry, ×100) (c).
DISCUSSION
Acromegaly is relatively rare with an incidence ranging from 3 to 11 per 1 million persons/year, and a prevalence of about 60/million.[
Occurring after fusion of the growth plates, individuals with acromegaly usually manifest with thickened bones of the jaw and extremities as well as hypertrophy of soft tissues giving the characteristic features of the disease, including coarse facial appearance, broadened nose, large hands and feet, obesity, organomegaly, hyperhidrosis, and snoring, the latter of which may be an early manifestation of obstructive sleep apnea. The present patient was positive for most of these features despite his preadolescent age. In considering acromegaly in this patient, it is important to consider the recent onset of snoring, rapid acral enlargement as evidenced by the significant increase in his shoe size, and recent enlargement of his lips and nasal wings. Nagata et al. reviewed GH-secreting pituitary adenomas in childhood and young adults and reported that no preadolescent patients had acromegalic features.[
Clinical suspicion is a critical factor in making a diagnosis of acromegaly in childhood.[
The role of anthropometric measurements in making a diagnosis of acromegaly in preadolescence has yet to be determined. Measurements, such as the heel pad thickness, ring size, lean body mass, fat mass, and bone mineral density, may be difficult to interpret and apply uniformly across the different age groups in the preadolescent stage. Whereas there are standardized cutoff points in the adult population, these cannot be accurately extrapolated and adapted for management in this younger population.[
In addition to clinical assessment, biochemical analysis and brain imaging contribute to making a diagnosis of acromegaly and confirming the presence of pituitary adenoma or hyperplasia. Hormonal assay is crucial for the diagnosis. The accuracy of random GH measurements in making a diagnosis of acromegaly has generally been questioned considering the pulsatile nature of GH secretion and its sensitivity to sleep. This is an even greater concern in children due to the additional changes in GH levels according to age.[
Genetic screening for markers of familial acromegaly is also an important aspect of management of acromegaly in preadolescence. About half of the cases of pituitary gigantism and acromegaly in young adults occur in the context of an identifiable germline or somatic mutation.[
The goals of treatment for acromegaly in children are amelioration of clinical signs and symptoms, prevention of comorbidities, and normalization of life expectancy as in adult patients.[
In preadolescence, medical therapy with somatostatin analogs, GH receptor antagonists, and dopamine agonists are reserved only for cases with uncontrollable disease after surgery. Pegvisomant, a GH-receptor antagonist, showed some promise in children in a small study, decreasing the serum IGF-1 levels with complete resolution of acromegalic features in all three children studied even when surgery, octreotide, and cabergoline had previously failed to produce satisfactory results.[
Radiotherapy carries a significant risk in the preadolescent population as it may result in panhypopituitarism with severe consequences for normal growth and development. Taking the delayed action of radiotherapy into consideration in addition to the complications arising from its use, it is rarely recommended in this population.[
The present patient has maintained normal random GH and IGF-1 levels after surgery, although his nadir GH in 75 g OGTT level showed a decrease from the preoperative level and has still not reached hormonal remission according to the adult criteria. Therapeutic efficacy is assessed using various indices and long-term follow-up should be adopted in this population, as the risk of recurrence may be slightly higher than in adults.[
CONCLUSIONS
This case illustrated the importance of high clinical suspicion in making a diagnosis of acromegaly even in younger patients. It is difficult, however, to achieve cure by resection through the EEA in preadolescent patients. Genetic testing and long-term follow-up with multidisciplinary management are recommended in such young patients.
Ethical approval
All procedures in studies involving human participants were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abe T, Tara LA, Lüdecke DK. Growth hormone-secreting pituitary adenomas in childhood and adolescence: Features and results of transnasal surgery. Neurosurgery. 1999. 45: 1-0
2. Blumberg DL, Sklar CA, David R, Rothenberg S, Bell J. Acromegaly in an infant. Pediatrics. 1989. 83: 998-1002
3. Burton T, Le Nestour E, Neary M, Ludlam WH. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary. 2016. 19: 262-7
4. Etxabe J, Gaztambide S, Latorre P, Vazquez JA. Acromegaly: An epidemiological study. J Endocrinol Invest. 1993. 16: 181-7
5. Goldenberg N, Racine MS, Thomas P, Degnan B, Chandler W, Barkan A. Treatment of pituitary gigantism with the growth hormone receptor antagonist pegvisomant. J Clin Endocrinol Metab. 2008. 93: 2953-6
6. Hannah-Shmouni F, Trivellin G, Stratakis CA. Genetics of gigantism and acromegaly. Growth Horm IGF Res. 2016. 30-31: 37-41
7. Johannsson G, Bidlingmaier M, Biller BMK, Boguszewski M, Casanueva FF, Chanson P. Growth hormone research society perspective on biomarkers of GH action in children and adults. Endocr Connect. 2018. 7: R126-R134
8. Lafferty AR, Chrousos GP. Pituitary tumors in children and adolescents. J Clin Endocrinol Metab. 1999. 84: 4317-23
9. Melmed S, Casanueva FF, Klibanski A, Bronstein MD, Chanson P, Lamberts SW. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013. 16: 294-302
10. Melmed S. Medical progress: Acromegaly. N Engl J Med. 2006. 355: 2558-73
11. Nagata Y, Inoshita N, Fukuhara N, Yamaguchi-Okada M, Nishioka H, Iwata T. Growth hormone-producing pituitary adenomas in childhood and young adulthood: Clinical features and outcomes. Pituitary. 2018. 21: 1-9
12. Petrossians P, Daly AF, Natchev E, Maione L, Blijdorp K, Sahnoun-Fathallah M. Acromegaly at diagnosis in 3173 patients from the Liège Acromegaly Survey (LAS) Database. Endocr Relat Cancer. 2017. 24: 505-18
13. Rajasoorya C, Holdaway IM, Wrightson P, Scott DJ, Ibbertson HK. Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol (Oxf). 1994. 41: 95-102
14. Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: Acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010. 72: 203-8
15. Renn WH, Rhoton AL Jr. Microsurgical anatomy of the sellar region. J Neurosurg. 1975. 43: 288-98
16. Steinbach HL, Russell W. Measurement of the heel-pad as an aid to diagnosis of acromegaly. Radiology. 1964. 82: 418-23
17. Todd RM. Acromegaly in a girl of 8 years. Arch Dis Child. 1958. 33: 49-54

