

- Department of Neurosurgery, Hospital 12 de Octubre, Madrid, Spain.
- Department of Oral and Maxillofacial Surgery Hospital 12 de Octubre, Madrid, Spain.
- Department of Radiology, Hospital 12 de Octubre, Madrid, Spain.
- Department of Craniofacial Unit (ERN CRANIO), Hospital 12 de Octubre, Madrid, Spain.
Correspondence Address:
Pablo M. Munarriz
Department of Oral and Maxillofacial Surgery Hospital 12 de Octubre, Madrid, Spain.
Department of Craniofacial Unit (ERN CRANIO), Hospital 12 de Octubre, Madrid, Spain.
DOI:10.25259/SNI_413_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Pablo M. Munarriz1,4, Beatriz Pascual1,4, Ana M. Castaño-Leon1,4, Ignacio García-Recuero2,4, Marta Redondo2,4, Ana Martínez de Aragón3,4, Ana Romance2,4. Apert syndrome: Cranial procedures and brain malformations in a series of patients. 29-Oct-2020;11:361
How to cite this URL: Pablo M. Munarriz1,4, Beatriz Pascual1,4, Ana M. Castaño-Leon1,4, Ignacio García-Recuero2,4, Marta Redondo2,4, Ana Martínez de Aragón3,4, Ana Romance2,4. Apert syndrome: Cranial procedures and brain malformations in a series of patients. 29-Oct-2020;11:361. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=10362
Date of Submission
07-Jul-2020
Date of Acceptance
28-Sep-2020
Date of Web Publication
29-Oct-2020
Abstract
Background: Apert syndrome is one of the most severe craniofacial disorders. This study aims to describe the craniofacial surgeries and central nervous system malformations of a cohort of children with Apert syndrome treated in the past 20 years and to compare these data with previously published data.
Methods: Retrospective analysis of a series of patients with Apert syndrome treated between 1999 and 2019 in our hospital. Information was analyzed regarding craniofacial procedures, hydrocephalus and presence of shunts, Chiari malformation Type 1, and other brain malformations such as corpus callosum and septum pellucidum anomalies.
Results: Thirty-seven patients were studied. Ventriculoperitoneal shunt prevalence was 24.3%, and 8.1% of patients required decompressive surgery for Chiari malformation. All of them needed at least one cranial vault remodeling procedure. The median age for this procedure was 8 months. In 69.7% of patients, the first cranial vault intervention was performed in the fronto-orbital region. In 36.4% of patients, a midface advancement had been performed at the time of this review, although this proportion was very dependent on the follow-up period and the age of the patients. The median age for the midface advancement procedure was 5.25 years. Anomalies of the corpus callosum and the septum pellucidum were reported in 43.2% and 59.5% of patients, respectively.
Conclusion: Apert syndrome is a type of syndromic craniosynostosis, and patients usually require one or more cranial and facial surgeries. In comparison with other syndromic craniosynostosis types, Apert syndrome less frequently requires a VP shunt or treatment for a Chiari malformation.
Keywords: Apert syndrome, Craniosynostosis, Fronto-orbital advancement, Hydrocephalus, Monobloc advancement
INTRODUCTION
Apert syndrome is a severe craniofacial syndrome that was initially described in 1906 by French physician Eugene Apert.[
Patients present with malformations related to the head, face, and limbs. At the skull, it is characteristic of bilateral coronal synostosis, although other sutures may be affected, and coronal sutures are rarely unaffected. Skull base development is abnormal, and the skull shape confers a steep and flattened forehead with a flat occiput. Midfacial hypoplasia and exorbitism are also characteristic of the disorder. Many patients have an associated cleft palate or bifid uvula.[
This study seeks to (1) collect and describe the craniofacial procedures performed in our series of Apert patients in the past 20 years; (2) assess the proportion of shunted patients and patients with Chiari malformation type 1 (CM1) needing surgery; and (3) describe a number of brain and cranial malformations that frequently occur in this disorder.
MATERIALS AND METHODS
Patients
We conducted a retrospective analysis of consecutive patients with Apert syndrome treated at Hospital 12 de Octubre, Madrid, Spain; in our Craniofacial Unit, between 1999 and 2019. Informed consents are obtained from parents regarding images and information database. Our database including clinical records and radiological records was examined, and the type, number, and order of surgeries were extracted from the database. The brain malformations associated with this disorder were analyzed using magnetic resonance imaging (MRI), and every patient underwent at least one MRI. The follow-up period was calculated and described as patients still attending the craniofacial clinic or those who were lost to follow-up.
Information of this series of patients was extracted regarding the following:
Incidence of hydrocephalus requiring ventriculoperitoneal shunt compared with a normal ventricular size or enlarged ventricular size without shunt (nonprogressive ventriculomegaly). Indications to perform VP shunt procedures were retrieved Incidence of CM1 defined as tonsillar herniation of 5 mm or greater. Proportion of patients requiring surgery and indication of such procedures Cranial and brain malformations detected through MRI studies Craniofacial surgical procedures: number, type, order, and age of the patient at the time of surgery.
Craniofacial surgeries
Various cranial vault procedures are performed in Apert patients and may be divided into surgeries of the anterior half of the skull or the posterior half, although one could even reach two-thirds of the skull. A total cranial vault remodeling or holocranial dismantling is also possible. With regard to the anterior half of the cranium, a remodeling of the frontal region can be performed with or without a supraorbital bandeau. If an advancement of the supraorbital rim and the frontal bones is instead performed, then it is called a fronto-orbital advancement (FOA). The advancement may be static (with absorbable or nonabsorbable miniplates), but it may also be progressive using distractors (osteogenic distraction). Regarding osteogenic distraction, our team has used a technique described by Hirabayashi et al.,[
On the other hand, patients may need surgical procedures affecting the face. Due to midfacial hypoplasia and upper airway obstruction, a midface advancement is often performed. A midfacial advancement may be performed alone (Le Fort III osteotomy) or in combination with a FOA (monobloc advancement).
RESULTS
We identified a total of 44 patients with Apert syndrome from the past two decades in our database. However, seven patients were excluded from analysis since the clinical information was scarce or they were treated mainly in a different hospital. Of the 37 remaining patients, 33 have been treated in our center from birth; thus, we have a complete information and surgical record. The remaining four patients were initially operated on in different centers and subsequently in ours. We included these four patients for analysis of hydrocephalus and VP shunt, Chiari malformation, and brain malformations (total: 37 patients), since no VP shunts or Chiari decompressions correspond to these four patients. However, we decided to not consider craniofacial surgeries since the indications were made outside our Craniofacial Center (total: 33 patients).
The 37 patients were comprised of 17 females (46%) and 20 males (54%) [
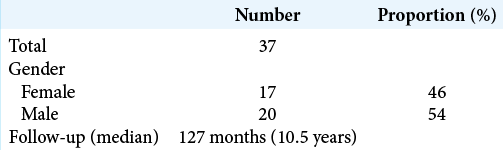
Hydrocephalus and ventricular size
Nine out of 37 patients (24.3%) required a VP shunt. For the rest, the ventricular size was categorized using the Evans’ index, that is, the ratio of maximum width of the frontal horns of the lateral ventricles and the maximal internal diameter of the skull at the same level (where >0.30 is considered ventriculomegaly). In this manner, nonshunted patients were categorized in two groups: those with normal ventricular size and those with ventriculomegaly (referred in the literature as “nonprogressive ventriculomegaly”). Eighteen patients (48.7%) presented a normal ventricular size, with an Evans’ index from 0.15 to 0.28. Ten patients (27%) presented nonprogressive ventriculomegaly, with an index oscillating from 0.31 to 0.44, and they did not present clinical findings related to intracranial hypertension and thus were not shunted. The total number of patients with shunt (hydrocephalus) and ventriculomegaly was 19 (51.3%).
Of the nine shunted patients, only one case was the shunt performed before any craniofacial procedure. In this case, the patient presented progressive hydrocephalus from birth and required shunting at 2 months of age. The remaining eight patients required a VP shunt after surgical procedures. The indication was CSF fistula in one patient and progressive hydrocephalus in seven patients. Of these seven patients, cranial vault remodeling surgeries preceded the VP shunt. Four were anterior half remodeling surgeries, two were posterior half remodeling surgeries, and one was a total cranial vault remodeling surgery. This proportion is not informative since the distribution of anterior/posterior/ holocranial vault remodeling in the complete series is very similar. Of the nine shunted patients, three patients needed a shunt revision (two patients twice and one patient once); additionally, one patient presented a shunt infection resolved with shunt replacement. In our unit, shunts for ApertCrouzon-Pfeiffer patients are placed through a posterior (occipital) burr hole to avoid potential contamination of an anterior shunt if they need halo placement for a midfacial advancement.
Chiari malformation
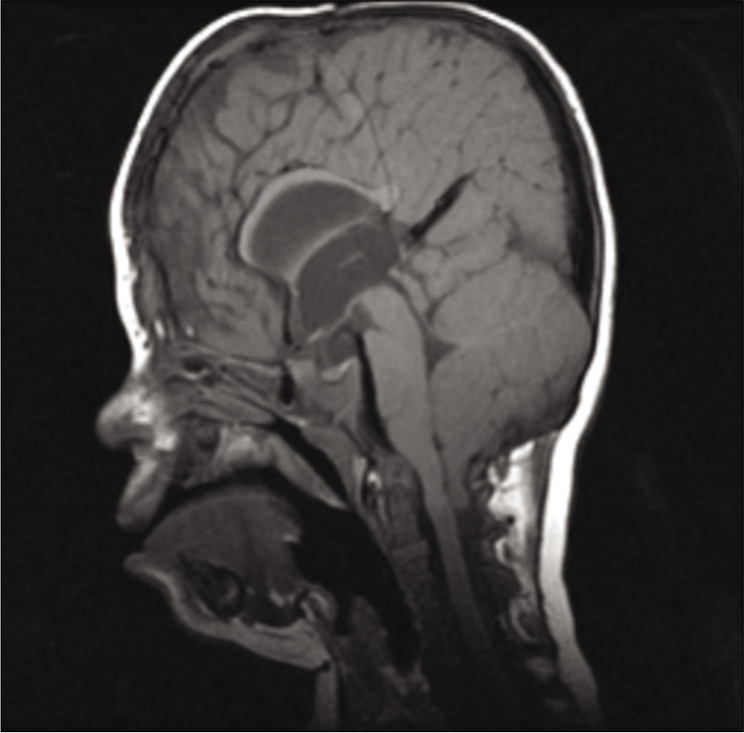
Cerebellar tonsillar herniation through the foramen magnum or CM1 was present in 8 out of 37 patients of our series (21.6%). Three patients required surgery (8.1%) consisting of a posterior fossa craniectomy with resection of the posterior arch of the C1 vertebrae, plus a wide dural opening and duroplasty. Two patients did not present any attributable symptom at the time of the procedure, and therefore, the indication was made on the basis of very severe radiological tonsillar herniation (>20 mm). One patient was 2 years old at the time, and the other was 3 years old. One patient required Chiari decompression at 9 months of age due to attributable symptoms such as progressive central sleep apneas in a tracheostomized patient. The remaining 5 patients (13.5%), despite presenting radiological diagnosis of Chiari malformation, did not present symptoms, and the tonsillar herniation was not considered sufficiently severe to indicate decompressive surgery. Most of the Apert patients within our series do not present Chiari malformation (29 patients, 78.4%).
Brain and cranial malformations on MRI studies
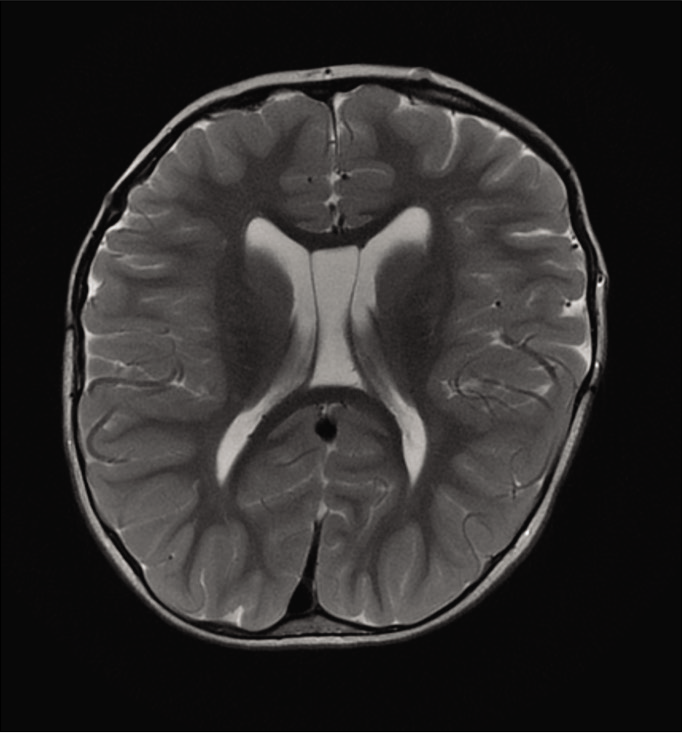
Thirty-seven patients were included since complete cranial MRI images were available. The majority of brain malformations related to Apert syndrome corresponded to abnormalities of midline development, more specifically involving the olfactory-limbic-septal-callosal structures. Sixteen patients presented corpus callosum malformations: 15 (40.5%) presented segmental or global hypoplasia and 1 (2.7%) presented complete agenesis. Septum pellucidum was analyzed, and complete or partial absence was found in 13 patients (35.2%), whereas 9 patients (24.3%) presented a double septum (cystic septum pellucidum or cavum vergae).
The amygdala and hippocampal region were also scrutinized. Two patients (5.4%) presented verticalized and malrotated hippocampus bilaterally, and one patient presented with intractable epilepsy and an amygdalar tumor that was resected and whose pathology revealed ganglioglioma. The petrous bone was also assessed and revealed nearly constant malformations in the inner ear. In 33 patients (89.2%), a dilated cystic vestibule was found. Craniocervical junction abnormalities were also common. In 30 patients (81.1%), remodeling of the clivus was confirmed, and 9 patients (24.3%) presented basilar invagination.
Craniofacial surgical procedures
The 33 patients treated from birth in our center were included in this section.
First surgical intervention
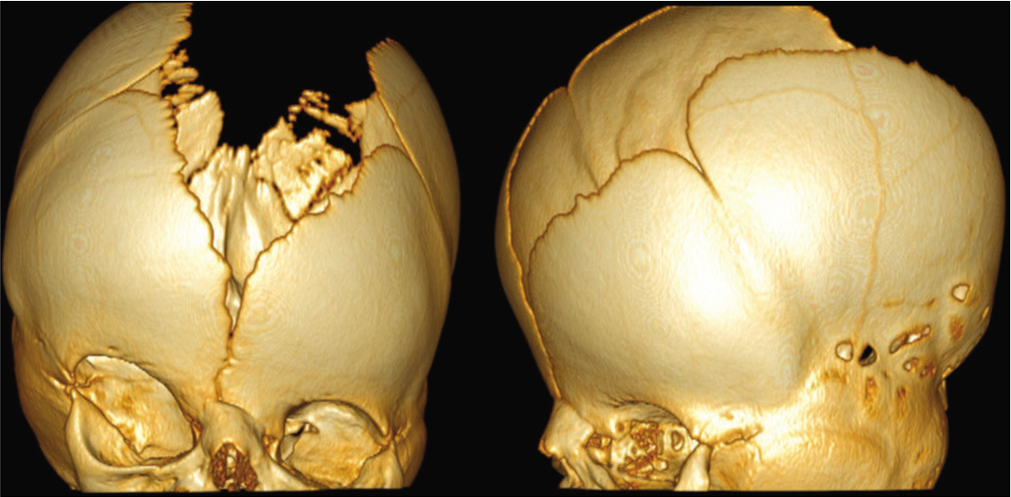
The first surgical intervention for 29 patients was a cranial vault procedure, whereas it was mandibular distraction in three patients and VP shunt in one patient. Every patient required a cranial vault surgery at least once. However, the first surgery involved the anterior half (fronto-orbital) in 23 patients (69.7%), the posterior half in 8 patients (24,2%), and a total cranial vault remodeling was carried out in 2 patients (6.1%). The median age for the first cranial vault surgery was 8 months (range 3–15 months), and it was performed before 6 months in 7 patients, between 6 and 12 months in 23 patients, and after 12 months of age in 3 patients.
Type of cranial procedures
A total of 53 cranial vault procedures were performed, including 6 monobloc advancements (frontal and facial). In all 33 patients, a FOA was performed at some point (2 of them as part of holocranial remodeling). Of them, 17 required only one cranial vault surgery. In this subgroup, 15 patients required an FOA, and two patients required a holocranial remodeling.
In 14 patients, two cranial vault procedures were performed. The combinations included FOA and posterior remodeling (7 patients), two FOAs (3 patients), FOA and monobloc advancement (3 patients), and posterior remodeling and monobloc advancement (1 patient). Finally, two patients required more than 2 cranial procedures. One patient underwent three procedures (two FOAs and a posterior remodeling), and another required five cranial vault surgeries (one holocranial remodeling, two FOAs, and two monobloc advancements).
Midfacial advancement and monobloc advancement
Of the 33 patients, at the time of this review, 12 of them required surgical treatment of their midfacial hypoplasia. This proportion was dependent on the age of the patients in the cohort, since nonoperated patients may require these procedures as they age. In fact, in an additional four patients, the midfacial advancement was indicated in outpatient clinic visits and remains pending. Of the 12 operated patients, 9 required only one procedure and 3 required more than 1. Sixteen procedures were undertaken, including 10 midfacial advancements (Le Fort III) and 6 monobloc advancements. The median age for the first procedure was 63 months (5.25 years). In six patients, the procedures were carried out between 5 and 10 years of age. In two patients, they were performed between 10 and 16 years of age, and in four patients, their first procedure was performed when they were <5 years of age.
DISCUSSION
The usual shape of the skull in patients affected of Apert syndrome is brachyturricephaly. Typically, the head width and height are increased, whereas the head length is reduced.[
CNS malformations
Apert syndrome is related with brain malformations that may be classified as primary or secondary to the osseous deformity.[
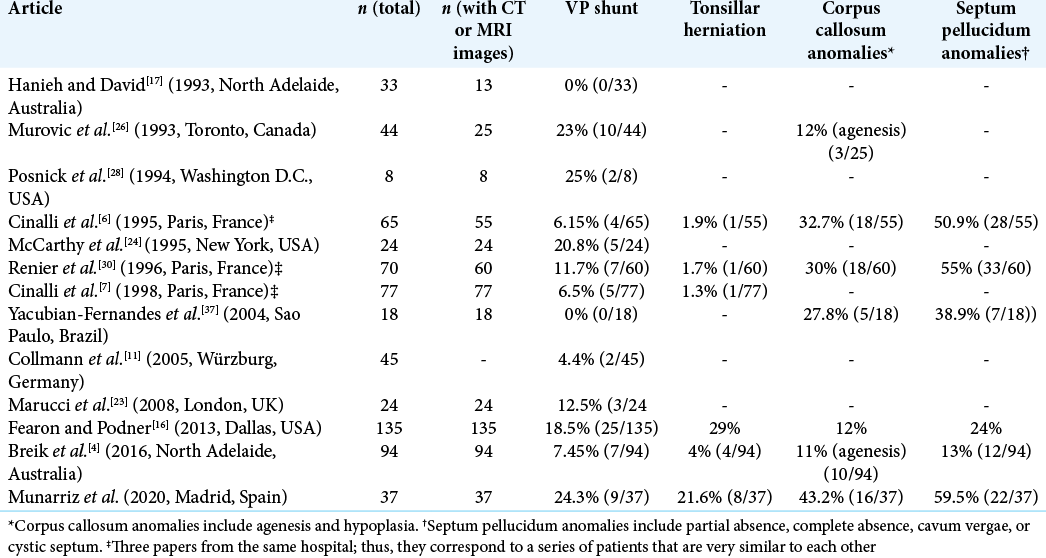
Another type of primary malformations comprises disorders of brain development itself. This type has been described as a greater frequency of abnormalities of midline development (olfactory-limbic-septal-callosal).[
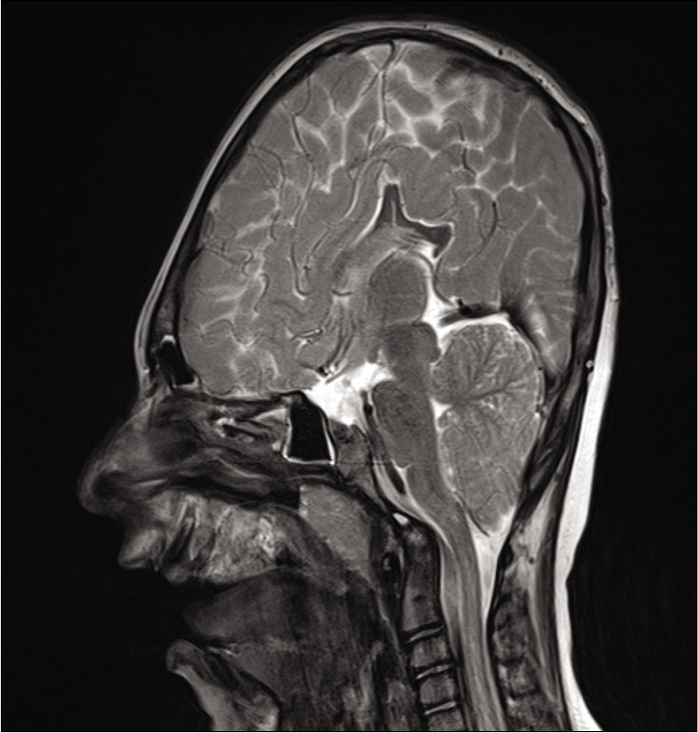
The main secondary malformation is cerebellar tonsillar herniation through the foramen magnum or Chiari type 1 malformation [
Craniofacial surgical procedures
The traditional paradigm of treatment in patients affected with Apert syndrome is to assume that they eventually require at least one surgery of cranial vault expansion or remodeling and thus to proceed with it during the 1st year of life. It is known that a fair number of patients will need more than 1 procedure regarding the skull region.[
From the paper of Fearon and Podner from the Craniofacial Center in Dallas,[
In the majority of units, however, the usual management has been to believe that a cranial vault procedure is needed and so to schedule it sometime within the 1st year of life. The controversy for the greater number of authors has been when to perform the first surgery.[
CONCLUSION
Apert syndrome is one of the most severe craniofacial disorders. Most will require a cranial vault surgical intervention, and many will require more than 1. The need for a midfacial advancement is also very prevalent. The incidence of hydrocephalus and the requirement of a VP shunt – as well as tonsillar herniation – are less common than in syndromes such as Crouzon or Pfeiffer.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We wish to acknowledge the tremendous contributions in treating our patients from several former members of our Craniofacial Unit (J. Hinojosa, M.J. Muñoz, J. Esparza).
References
1. Allam KA, Wan DC, Khwanngern K, Kawamoto HK, Tanna N, Perry A. Treatment of apert syndrome: A long-term follow-up study. Plast Reconstr Surg. 2011. 127: 1601-11
2. Anderson PJ, Netherway DJ, Abbott AH, Cox T, Roscioli T, David DJ. Analysis of intracranial volume in apert syndrome genotypes. Pediatr Neurosurg. 2004. 40: 161-4
3. Breik O, Mahindu A, Moore MH, Molloy CJ, Santoreneos S, David DJ. Apert syndrome: Surgical outcomes and perspectives. J Craniomaxillofac Surg. 2016. 44: 1238-45
4. Breik O, Mahindu A, Moore MH, Molloy CJ, Santoreneos S, David DJ. Central nervous system and cervical spine abnormalities in apert syndrome. Childs Nerv Syst. 2016. 32: 833-8
5. Carrera IA, Martínez-Frías ML, Pérez JJ, Grisolía LP, Rodríguez AC, Conde CN. Apert syndrome: Clinicoepidemiological analysis of a series of consecutive cases in Spain. An Esp Pediatr. 1999. 51: 667-72
6. Cinalli G, Renier D, Sebag G, Sainte-Rose C, Arnaud E, Pierre-Kahn A. Chronic tonsillar herniation in crouzon’s and apert’s syndromes: The role of premature synostosis of the lambdoid suture. J Neurosurg. 1995. 83: 575-82
7. Cinalli G, Sainte-Rose C, Kollar EM, Zerah M, Brunelle F, Chumas P. Hydrocephalus and craniosynostosis. J Neurosurg. 1998. 88: 209-14
8. Cohen MM, Kreiborg S. A clinical study of the craniofacial features in apert syndrome. Int J Oral Maxillofac Surg. 1996. 25: 45-53
9. Cohen MM, Kreiborg S. An updated pediatric perspective on the apert syndrome. Am J Dis Child. 1993. 147: 989-93
10. Cohen MM, Kreiborg S. The central nervous system in the apert syndrome. Am J Med Genet. 1990. 35: 36-45
11. Collmann H, Sörensen N, Krauss J. Hydrocephalus in craniosynostosis: A review. Childs Nerv Syst. 2005. 21: 902-12
12. David DJ, Anderson P, Flapper W, Syme-Grant J, Santoreneos S, Moore M. Apert syndrome: Outcomes from the australian craniofacial unit’s birth to maturity management protocol. J Craniofac Surg. 2016. 27: 1125-34
13. Davis C, MacFarlane MR, Wickremesekera A. Occipital expansion without osteotomies in apert syndrome. Childs Nerv Syst. 2010. 26: 1543-8
14. de León GA, de León G, Grover WD, Zaeri N, Alburger PD. Agenesis of the corpus callosum and limbic malformation in apert syndrome (Type I acrocephalosyndactyly). Arch Neurol. 1987. 44: 979-82
15. Esparza J, Hinojosa J, García-Recuero I, Romance A, Pascual B, de Aragón AM. Surgical treatment of isolated and syndromic craniosynostosis. Results and complications in 283 consecutive cases. Neurocirugia (Astur). 2008. 19: 509-29
16. Fearon JA, Podner C. Apert syndrome: Evaluation of a treatment algorithm. Plast Reconstr Surg. 2013. 131: 132-42
17. Hanieh A, David DJ. Apert’s syndrome. Childs Nerv Syst. 1993. 9: 289-91
18. Hirabayashi S, Sugawara Y, Sakurai A, Harii K, Park S. Frontoorbital advancement by gradual distraction. Technical note. J Neurosurg. 1998. 89: 1058-61
19. Hoffman HJ, Hendrick EB. Early neurosurgical repair in craniofacial dysmorphism. J Neurosurg. 1979. 51: 796-803
20. Hogan GR, Bauman ML. Hydrocephalus in apert’s syndrome. J Pediatr. 1971. 79: 782-7
21. Kreiborg S, Aduss H, Cohen MM Jr. Cephalometric study of the apert syndrome in adolescence and adulthood. J Craniofac Genet Dev Biol. 1999. 19: 1-11
22. Marsh JL, Galic M, Vannier MW. Surgical correction of the craniofacial dysmorphology of apert syndrome. Clin Plast Surg. 1991. 18: 251-75
23. Marucci DD, Dunaway DJ, Jones BM, Hayward RD. Raised intracranial pressure in apert syndrome. Plast Reconstr Surg. 2008. 122: 1162-8
24. McCarthy JG, Glasberg SB, Cutting CB, Epstein FJ, Grayson BH, Ruff G. Twenty-year experience with early surgery for craniosynostosis: II. The craniofacial synostosis syndromes and pansynostosis-results and unsolved problems. Plast Reconstr Surg. 1995. 96: 284-95
25. Mulliken JB, Bruneteau RJ. Surgical correction of the craniofacial anomalies in apert syndrome. Clin Plast Surg. 1991. 18: 277-89
26. Murovic JA, Posnick JC, Drake JM, Humphreys RP, Hoffman HJ, Hendricks EB. Hydrocephalus in apert syndrome: A retrospective review. Pediatr Neurosurg. 1993. 19: 151-5
27. Noetzel MJ, Marsh JL, Palkes H, Gado M. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985. 107: 885-92
28. Posnick JC, Lin KY, Jhawar BJ, Armstrong D. Apert syndrome: Quantitative assessment by CT scan of presenting deformity and surgical results after first-stage reconstruction. Plast Reconstr Surg. 1994. 93: 489-97
29. Raybaud C, di Rocco C. Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: A review. Childs Nerv Syst. 2007. 23: 1379-88
30. Renier D, Arnaud E, Cinalli G, Sebag G, Zerah M, Marchac D. Prognosis for mental function in apert’s syndrome. J Neurosurg. 1996. 85: 66-72
31. Renier D, Lajeunie E, Arnaud E, Marchac D. Management of craniosynostoses. Childs Nerv Syst. 2000. 16: 645-58
32. Spruijt B, Rijken BF, den Ottelander BK, Joosten KF, Lequin MH, Loudon SE. First vault expansion in apert and crouzon-pfeiffer syndromes: Front or back?. Plast Reconstr Surg. 2016. 137: 112e-21e
33. Tan AP, Mankad K. Apert syndrome: Magnetic resonance imaging (MRI) of associated intracranial anomalies. Childs Nerv Syst. 2018. 34: 205-16
34. Whitaker LA, Bartlett SP, Schut L, Bruce D. Craniosynostosis: An analysis of the timing, treatment, and complications in 164 consecutive patients. Plast Reconstr Surg. 1987. 80: 195-212
35. Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD. Apert syndrome results from localized mutations of FGFR2 and is allelic with crouzon syndrome. Nat Genet. 1995. 9: 165-72
36. Wong GB, Kakulis EG, Mulliken JB. Analysis of fronto-orbital advancement for apert, crouzon, pfeiffer, and saethre-chotzen syndromes. Plast Reconstr Surg. 2000. 105: 2314-23
37. Yacubian-Fernandes A, Palhares A, Giglio A, Gabarra RC, Zanini S, Portela L. Apert syndrome: Analysis of associated brain malformations and conformational changes determined by surgical treatment. J Neuroradiol. 2004. 31: 116-22

