

- Department of Neurosurgery, Jessa Hospital, Hasselt, Belgium
- Department of Neurosurgery, ZAS Augustinus, Antwerp, Belgium
- Department of Neurosurgery, St. Trudo Hospital, Sint-Truiden, Belgium
- Department of Neurosurgery, St. Franciscus Hospital, Heusden-Zolder, Belgium
- Department of Study and Educational Center for Neurosurgery, Virga Jesse, Belgium
- Faculty of Medicine and Life Science, Hasselt University, Hasselt, Belgium
Correspondence Address:
Leonie Witters, Department of Neurosurgery, ZAS Augustinus, Wilrijk, Belgium.
DOI:10.25259/SNI_1132_2024
Copyright: © 2025 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Leonie Witters1,2, Salah-Eddine Achahbar1,3,4,5, Samuel Klein1,3,4,5, Sacha Meeuws1,3,4,5, Mark Plazier1,3,4,5,6, Eric Put1,3,4,5, Gert Roosen1,3,4,5, Steven Vanvolsem1,3,4,5, Maarten Wissels1,3,4,5, Sven Bamps1,3,4,5. Chiari 1 malformation in patient with Noonan syndrome: A case report and review of literature. Surg Neurol Int 11-Apr-2025;16:132
How to cite this URL: Leonie Witters1,2, Salah-Eddine Achahbar1,3,4,5, Samuel Klein1,3,4,5, Sacha Meeuws1,3,4,5, Mark Plazier1,3,4,5,6, Eric Put1,3,4,5, Gert Roosen1,3,4,5, Steven Vanvolsem1,3,4,5, Maarten Wissels1,3,4,5, Sven Bamps1,3,4,5. Chiari 1 malformation in patient with Noonan syndrome: A case report and review of literature. Surg Neurol Int 11-Apr-2025;16:132. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=13495
Date of Submission
31-Dec-2024
Date of Acceptance
14-Mar-2025
Date of Web Publication
11-Apr-2025
Abstract
BackgroundDifferent theories exist about the pathogenesis of Chiari 1 malformations (CM-I), but none of them is thought to be exhaustive. Likewise, the role of genetic factors contributing to these conditions has not yet been elucidated, but there is a co-occurrence of CM-I with genetic syndromes such as Noonan syndrome (NS) and other RASopathies.
Case DescriptionWe describe the case of a 16-year-old female known with NS, currently presenting with Valsalva-induced headaches. Imaging of the brain and spine showed a CM-I with extensive syringohydromyelia. The patient was treated with a foramen magnum decompression and C1 laminectomy with duraplasty. The postoperative course was uneventful and the symptoms improved postoperatively.
ConclusionIn the literature, sixteen cases of CM-I in patients with NS are reported. Our reported case illustrates the co-occurrence between CM-I and RASopathies. We review current literature about the understanding of the possible association or pathogenetic link between the two conditions. This case report highlights the clinical importance of recognizing the co-occurrence of CM-I and NS, potentially guiding early diagnosis and management strategies.
Keywords: Chiari decompression, Chiari I malformation, Noonan syndrome, RASopathy
INTRODUCTION
Chiari 1 malformations (CM-I) are a group of disorders characterized by herniation of the cerebellar tonsils through the foramen magnum. Different theories about the pathogenesis of CM-I exist, but none of them is thought to be exhaustive.[
NS is a part of the family of RASopathies, a group of genetic conditions caused by mutation in genes encoding for proteins in the Ras/MAPK-signaling pathway. It is characterized by facial deformities, cardiovascular abnormalities, and thoracic deformities, but other organ systems can also be involved.[
The exact link between CM-I and NS is not completely elucidated. It is, however important to be aware of a possible association between CM-I and NS. In the management of patients with NS, clinicians have to be vigilant for neurological complaints caused by CM-I. When treating patients with CM-I, clinicians should start the appropriate diagnostic workup when features of NS are encountered.
In this article, we report the case of a 16-year-old female known with NS and diagnosed with CM-I. Furthermore, we discuss the current evidence on the association between the two disorders.
CASE DESCRIPTION
We report the case of a 16-year-old female diagnosed with NS at infantile age as a result of aberrant facial characteristics. The diagnosis was supported by genetic testing, showing a PTPN11 mutation compatible with NS.
She presented to the neurosurgical department with complaints of Valsalva-induced headaches. Upon clinical examination, we noticed that the patient was small for her age and displayed classical morphological features, as seen in NS. Neurological examination showed a symmetrical hyperreflexia in the four limbs without the presence of pathological reflexes. Further neurological testing was normal.
A magnetic resonance imaging scan of the brain showed a CM-I with the descent of the cerebellar tonsils of thirteen millimeters below the foramen magnum [
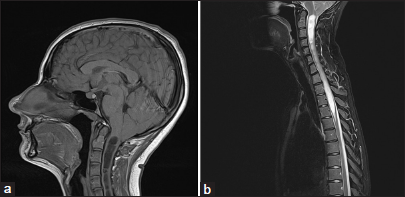
Taking into account the patient’s complaints, with a typical Valsalva-induced headache and the iconographic findings, with a CM-I with extensive syrinx, a surgical treatment was proposed.
The patient underwent a foramen magnum decompression with C1 laminectomy and duraplasty. Preoperative coagulation tests were normal, and no specific measurements were taken perioperatively. The procedure was uneventful, and the postoperative recovery proceeded without complications. During follow-up, the patient reported an improvement in headaches and decreased need for analgesics. One year after the surgery, the patient continues to be free of any complaints.
DISCUSSION
CM-I is a group of disorders characterized by a descent of the cerebellar tonsils through the foramen magnum of more than 5 mm in adults and 3 mm in children, possibly associated with syringomyelia. The diagnosis can be incidental in asymptomatic cases or can be prompted by symptoms. Classical symptoms are a Valsalva-induced occipital headache or symptoms due to brain stem compression, like central sleep apnea.[
Different theories about the pathogenesis of Chiari malformations have been described.[
RAS genes are involved in the Ras/mitogen-activated protein kinase (MAPK)-pathway.[
NS is a heterogeneous and multisystemic genetic disorder first described by Jacqueline Noonan in 1962. It is a relatively common condition with an estimated prevalence of 1 in 1000 to 1 in 2500 live births.[
NS is characterized by distinct facial and musculoskeletal features, which most often lead to the diagnosis of NS. The typical facial features include a tall forehead, hypertelorism, epicanthal folds, ptosis, and low-set posteriorly rotated ears. Musculoskeletal features are short stature, a short neck with low posterior hairline, a chest deformity with superior pectus carinatum and inferior pectus excavatum, and scoliosis. In over 80% of patients with NS, cardiovascular disorders can be found. The most commonly found disorder is pulmonary valve stenosis, but an atrium septum defect and hypertrophic cardiomyopathy are also frequent. Other manifestations of NS include a developmental delay, lymphatic issues, bleeding diathesis, and cryptorchidism.[
Few neurological disorders have been described in NS. Cognitive and developmental issues are reported more frequently in NS but are very variable. Structural disorders of the nervous system are rare. In literature, the presence of a CM-I in patients with NS is described in sixteen patients.[
In several studies, a co-occurrence of CM-I and RASopathies has been described. For example, in a paper reviewing 500 children who underwent surgery for CM-I, 5% had a diagnosis of neurofibromatosis type 1.[
It is often thought that NS is associated with CM-I because of the associated skull deformities. NS primarily influences facial features.[
To manage CM-I in patients with NS, an understanding of the natural history of CM-I is essential.[
The knowledge of the co-occurrence between RASopathies and CM-I is clinically relevant. First, it stimulates clinicians who regularly follow patients with RASopathies to remain vigilant for neurological symptoms or abnormalities during neurological examination and to perform appropriate imaging in a timely manner. Vice versa, it is a reminder to perform genetic work-up in patients with CM-I with possible features of RASopathies.
Ethical approval
This study is in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Ayaz E, Yildirim R, Celebi C, Ozalkak S. Noonan syndrome: Neuroimaging findings and morphometric analysis of the cranium base and posterior fossa in children. J Neuroimaging. 2023. 33: 318-27
2. Bamps S, Oyen T, Legius E, Vandenoord J, Stas M. Multiple granular cell tumors in a child with Noonan syndrome. Eur J Pediatr Surg. 2013. 23: 257-9
3. Buell TJ, Heiss JD, Oldfield EH. Pathogenesis and cerebrospinal fluid hydrodynamics of the Chiari I malformation. Neurosurg Clin N Am. 2015. 26: 495-9
4. Falls CJ, Page PS, Greeneway GP, Resnick DK, Stadler JA. Management of failed Chiari decompression and intrasyringeal hemorrhage in Noonan syndrome: Illustrative cases. J Neurosurg Case Lessons. 2022. 3: CASE21625
5. Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: Brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A. 2010. 152A: 1161-8
6. Han Y, Chen M, Wang H. Chiari I malformation in patients with RASopathies. Childs Nerv Syst. 2021. 37: 1831-6
7. Holder-Espinasse M, Winter RM. Type 1 Arnold-Chiari malformation and Noonan syndrome. A new diagnostic feature? Clin Dysmorphol 2003. ;. 12: 275
8. Kanodia G, Parihar V, Yadav YR, Bhatele PR, Sharma D. Morphometric analysis of posterior fossa and foramen magnum. J Neurosci Rural Pract. 2012. 3: 261-6
9. Keh YS, Abernethy L, Pettorini B. Association between Noonan syndrome and Chiari I malformation: A case-based update. Childs Nerv Syst. 2013. 29: 749-52
10. McClugage SG, Oakes WJ. The Chiari I malformation. J Neurosurg Pediatr. 2019. 24: 217-26
11. Nakamura T, Gulick J, Colbert MC, Robbins J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci U S A. 2009. 106: 11270-5
12. Noonan JA, Ehmke DA. Associated noncardiac malformations in children with congenital heart disease. Midwest Soc Pediatr Res. 1963. 63: 468-70
13. Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013. 14: 355-69
14. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013. 381: 333-42
15. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics. 2010. 126: 746-59
16. Saletti V, Vigano I, Melloni G, Pantaleoni C, Vetrano IG, Valentini LG. Chiari I malformation in defined genetic syndromes in children: Are there common pathways?. Childs Nerv Syst. 2019. 35: 1727-39
17. Samuels M, Northrup H. Noonan syndrome and type 1 Chiari malformation: Possible association. Am J Med Genet A. 2024. 194: e63776
18. Tubbs RS, Beckman J, Naftel RP, Chern JJ, Wellons JC, Rozzelle CJ. Institutional experience with 500 cases of surgically treated pediatric Chiari malformation Type I. J Neurosurg Pediatr. 2011. 7: 248-56
19. Yi Z, Xue J, Song Z, Li F, Yang C, Zhang Y. A patient with a PTPN11 gene variant complicated with Chiari I malformation and syringomyelia and a review of literatures. Int J Dev Neurosci. 2025. 85: e10396

