

- Department of Endocrinology-Diabetology and Nutrition, Medical School, Mohammed VI University Hospital Centre, Oujda, Oujda-Angad, Morocco.
- Department of Radiology, Medical School, Mohammed VI University Hospital Centre, Oujda, Oujda-Angad, Morocco.
- Department of Neurosurgery Medical School, Mohammed VI University Hospital Centre, Oujda, Oujda-Angad, Morocco.
- Department of Epidemiology, Medical School, Mohammed VI University Hospital Centre, Oujda, Oujda-Angad, Morocco.
DOI:10.25259/SNI_454_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Marouan Karrou1, Salma Benyakhlef1, Achwak Alla1, Najoua Messaoudi1, Asmae Oulad Amar2, Siham Rouf1, Imane Kamaoui2, Noureddine Oulali3, Faycal Moufid3, Naima Abda4, Hanane Latrech1. Clinical presentation and management of hypophysitis: An observational study of case series. 28-Jun-2021;12:304
How to cite this URL: Marouan Karrou1, Salma Benyakhlef1, Achwak Alla1, Najoua Messaoudi1, Asmae Oulad Amar2, Siham Rouf1, Imane Kamaoui2, Noureddine Oulali3, Faycal Moufid3, Naima Abda4, Hanane Latrech1. Clinical presentation and management of hypophysitis: An observational study of case series. 28-Jun-2021;12:304. Available from: https://surgicalneurologyint.com/surgicalint-articles/10932/
Date of Submission
08-May-2021
Date of Acceptance
24-May-2021
Date of Web Publication
28-Jun-2021
Abstract
Background: Hypophysitis is described as a rare chronic inflammatory affection of the pituitary gland. However, to date, its pathogenesis has not been completely cleared up. Clinical features are polymorphic, including symptoms related to inflammatory compression and/or hypopituitarism. Laboratory tests determine hormone deficiencies orientating replacement therapy’s protocol. MRI of the hypothalamic-pituitary region is crucial in exhibiting major radiological signs such as pituitary homogeneous enlargement and gland stalk’s thickening. The etiological diagnosis is still challenging without affecting the management strategy. Corticosteroids have widely been used but a close follow-up without any treatment has also been approved.
Case Description: In this report, seven patients with hypophysitis have been collected over a period of 6 years. The average age of our patients was 32.1 years ± 11.8 with a female predominance (71.4%). Panhypopituitarism was objective in 42.9% of cases, a combined deficiency of the hypothalamic-pituitary thyroid, adrenal and gonadal axes in 28.6% of cases. A central diabetes insipidus was noted in 42.9% of the patients. Idiopathic hypophysitis was the most common etiology. The use of long course corticosteroids was required in 28.6% when compressive signs were reported.
Conclusion: Hypophysitis remains a rare disease with nonspecific clinical and radiological patterns. Autoimmune origin seems to be the most frequent etiology. No guidelines have been established for hypophysitis management and the evolution is still unpredictable.
Keywords: Central diabetes insipidus, Corticosteroids, Hormone replacement, Hypophysitis, Hypopituitarism
INTRODUCTION
Hypophysitis is described as a rare chronic inflammatory affection of the pituitary gland, which may then damage the pituitary tissue and be responsible for temporary or permanent endocrine disorders.[
Hypophysitis classification is mainly based on histological and etiological patterns. Therefore, four histological types have been described, starting with the granulomatous hypophysitis described in 1917,[
Hypophysitis clinical features are polymorphic usually related to sella and parasella compression illustrated clinically by headache, nausea, vomiting and visual disturbances, pituitary hormone deficiencies, or diabetes insipidus.[
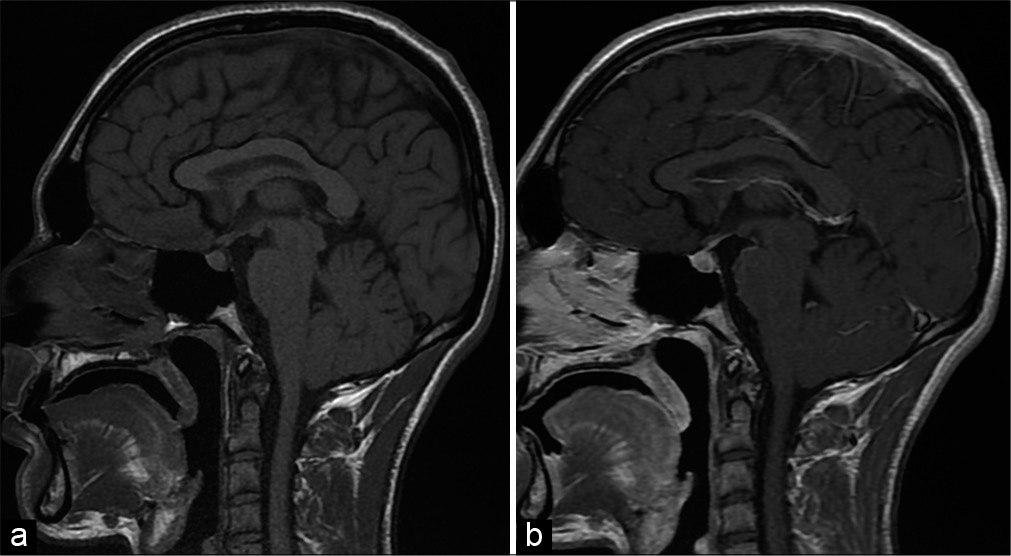
Several radiological signs may be very useful for diagnosis discussion, especially homogeneous pituitary enlargement with intense contrast enhancement and loss of bright spot of the neurohypophysis on T1-weighted images.[
Hypophysitis management revolves around the necessity of precocious hormone replacement therapy, the decision of corticosteroid treatment initiation, and the use of decompression surgery once we have the indication.
In this report, we describe seven cases of hypophysitis diagnosed through clinical symptoms and imaging patterns without pathology examination, with good evolution under replacement therapy and corticosteroids in one case. Regression of hypophysitis clinical and radiological signs was observed in most of our patients even without corticosteroids therapy.
CLINICAL AND PARACLINICAL EVALUATION
All of the patients included in this study [
RESULTS
Each patient underwent a clinical examination, besides the evaluation of both anterior and posterior pituitary functions. Patients consulted for signs of sella compression in 85.7% of cases (headache: 85.7%, vomiting: 71.4%, and visual disturbance in 57.1%) and a polyuria-polydipsia syndrome (PPS) in 42.9% of the cases.
Endocrine assessment of our patients showed a corticotropic, thyrotropic, and gonadotropic axis deficit. In 71%, the somatotropic axis was deficient in 28% of cases, however, hyperprolactinemia was observed in one patient. A panhypopituitarism in 42.9% of cases. A central diabetes insipidus was observed in 42.9% of the patients.
The most observed radiological features are homogeneous and symmetric pituitary enlargement with intense contrast enhancement in 57.1% [
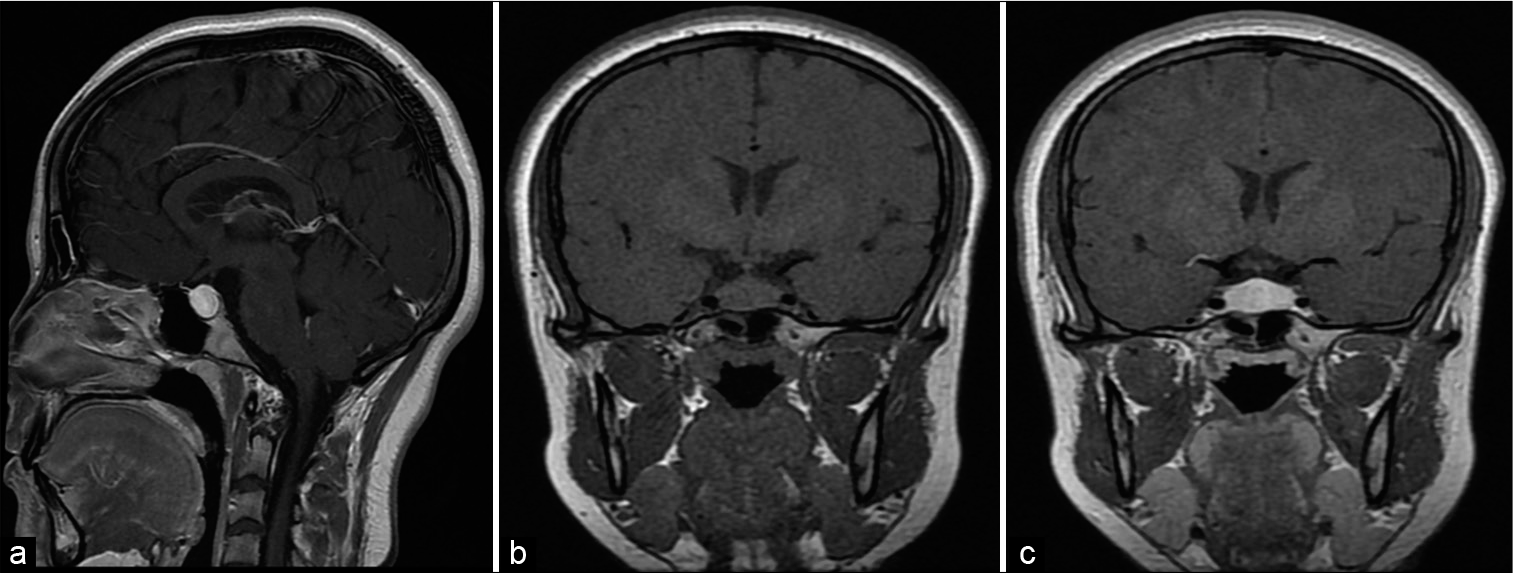
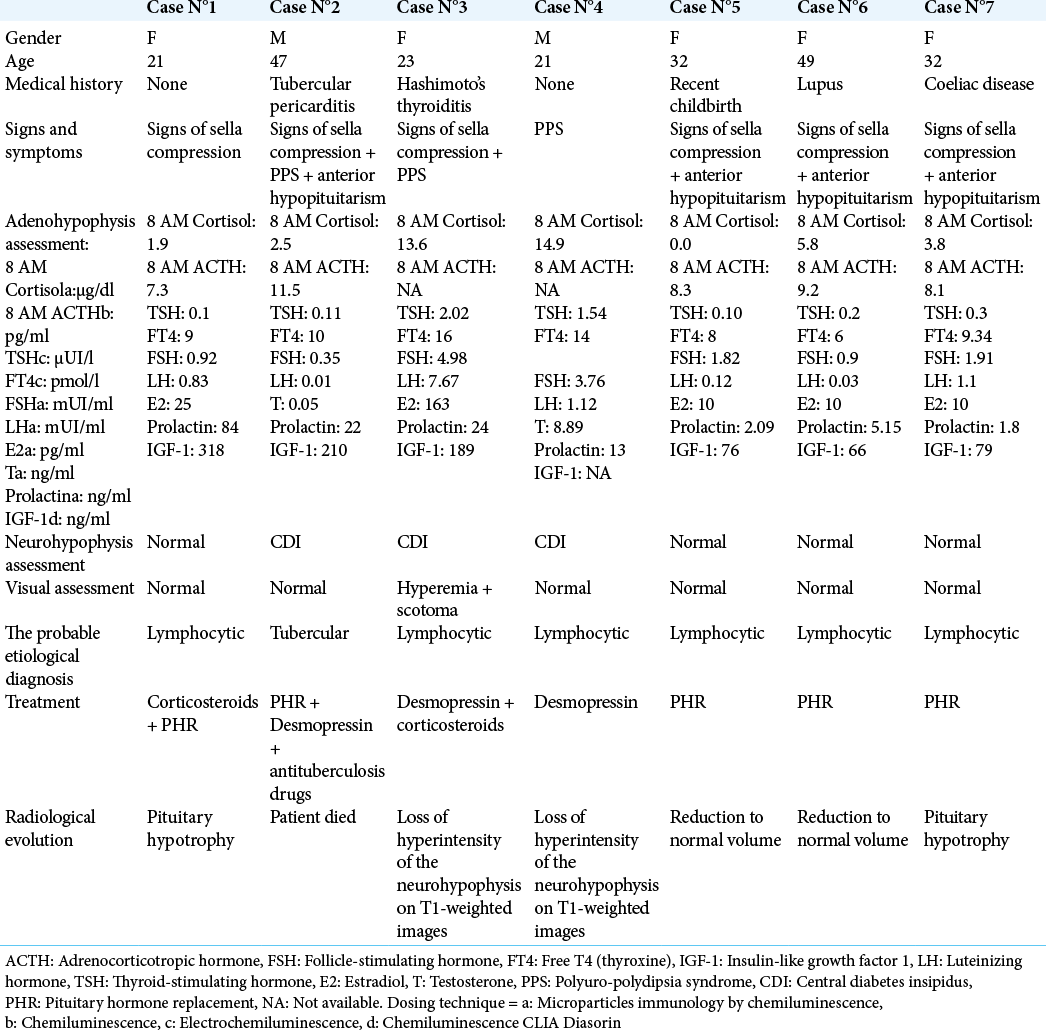
Figure 1:
Hypothalamic-pituitary MRI of patient number 1 before corticosteroid treatment, showing a homogeneous hypertrophy of the pituitary gland with high contrast after gadolinium injection. (a) Sagittal section SE T1 after gadolinium injection (swelling of the antehypophyseal gland with homogeneous enhancement after contrast). (b and c) Pituitary dynamic sequence in coronal section SE T1 before (b) and after (c) gadolinium injection.
The analysis of patients’ medical history disclosed an autoimmune disease in 42.9% of cases, a tuberculous pericarditis, and recent childbirth in 14.3% of cases. The etiological diagnosis was based on clinical, biological, and radiological criteria. A probable lymphocytic hypophysitis was noted in 85.7% of cases (adenohypophysitis in 50%, infundibulo-neurohypophysitis in 33.4%, and panhypophysitis in 16.7%) and tuberculosis in 14.4% of cases. Therapeutic management relied on the etiological diagnosis. The use of a long course corticosteroid therapy was required in 28.6% of subjects in case of visual disturbances due to compression, in addition to hormone replacement therapy (HYDROCORTISON and LEVOTHYROXIN in 71%, DYDROGESTERON + ESTRADIOL in 28.6%), and desmopressin in patients with central diabetes insipidus (42.85%).
Outcome and follow-up
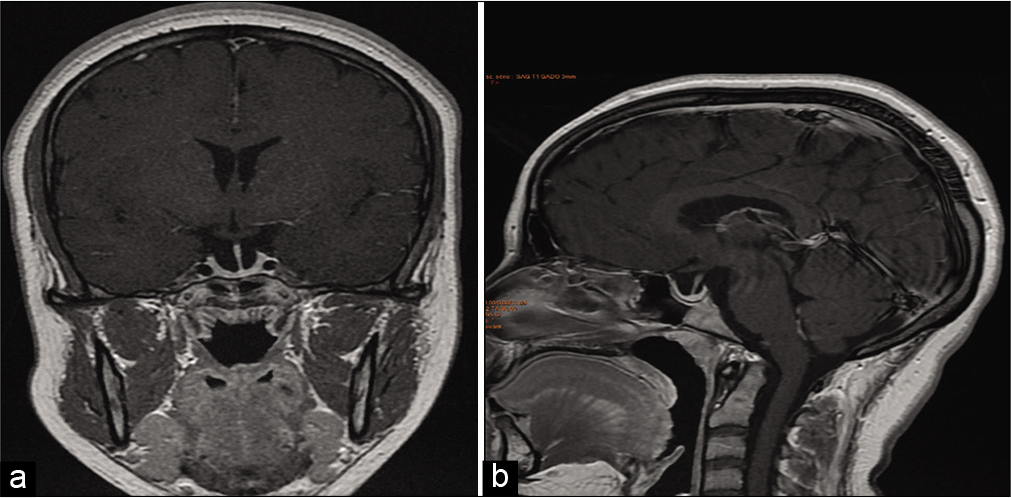
MRI was performed every 6 months, especially during the first 2 years, to assess the progression of the disease. Pituitary deficiencies were reassessed to adjust hormone dosing and monitor new endocrine disorders. Clinical improvement was achieved in all our patients with a reduction to normal volume in 2 cases (28.57%). The patients were kept under clinical observation. Pituitary hypotrophy was observed in one patient treated with glucocorticoids [Case 1,
DISCUSSION
The chronic inflammation of the pituitary gland is a rare condition, explaining the low number of reported cases in the literature. We have been able to compile seven cases over a period of 6 years, whereas Fedala et al. in Algeria reported a total of 15 cases over 16 years[
A clear predominance of women was noted by most authors throughout literature.[
Literature data[
Biological assessment of hypophysitis aimed to determine which axes are deficient and to confirm the etiology of the observed central diabetes insipidus responsible of a PPS. The number of axes involved is variously reported in literature depending on the authors. The adrenal axis seems to be the most involved followed by the thyroid axis and then the gonadal axis.[
The presence of a diabetes insipidus implies an autoimmune involvement of the infundibulo-neurohypophysis specifically the vasopressin-secreting cells, by anti-hypothalamic antibodies.[
MRI was an efficient imaging tool in the assessment of the hypothalamic-pituitary region. Radiological diagnosis of hypophysitis can sometimes tend to be difficult due to the polymorphic nature of the lesions, however, certain findings are of great orientation value. In particular, a pituitary enlargement [
The real challenge for an endocrinologist of a radiologist is to differentiate between a hypophysitis and its main differential, the holosellar pituitary adenoma. For that purpose, we use Gutenberg et al. radiological score[
Throughout literature, various etiologies were described by different authors. Our study reported a lymphocytic involvement in 80% of cases versus granulomatous involvement in 20% of cases in accordance with data from Guo et al. series.[
In the absence of any consensus regarding management, multiple therapeutic protocols were described throughout literature,[
In the literature review carried out by Karaca and Kelestimur,[
Right after the analysis of this study and the available data in the literature, we found that the evolution of hypophysitis was unpredictable and inconstant. In fact, it could be either favorable (clinical improvement, recovery of pituitary functions, and pituitary volume regression on MRI) under monitoring alone or under treatment, or disadvantageous with the persistence of clinical signs, deterioration of pituitary functions, recurrence, etc. Lupi et al. demonstrated in an experimental animal model demonstrating that the evolution of hypophysitis can lead to an empty sella [
CONCLUSION
Hypophysitis remains a rare disease with a polymorphic and nonspecific clinical and radiological presentation. Primary damage to the pituitary gland of autoimmune origin seems to be the most frequent etiology and its management should revolve around the need to correct hormonal deficits, and to remove the compression on neighboring structures if present. However, the evolution varies from patient to patient hence the need for multidisciplinary management.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Allix I, Rohmer V. Hypophysite: Un spectre étiologique de plus en plus large. ? Ann Endocrinol. 2012. 73: S17-25
2. Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R. The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol. 2014. 170: 161-72
3. Bonneville JF. Imagerie des hypophysites. Ann Endocrinol. 2012. 73: 76-7
4. Buxton N, Robertson I. Lymphocytic and granulocytic hypophysitis: A single centre experience. Br J Neurosurg. 2001. 15: 242-5
5. Caturegli P, Iwama S. From Japan with love: Another tessera in the hypophysitis mosaic. J Clin Endocrinol Metab. 2013. 98: 1865-8
6. Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008. 7: 631-7
7. Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005. 26: 599-614
8. De Bellis A, Sinisi AA, Pane E, Dello Iacovo A, Bellastella G, Di Scala G. Involvement of hypothalamus autoimmunity in patients with autoimmune hypopituitarism: Role of antibodies to hypothalamic cells. J Clin Endocrinol Metab. 2012. 97: 3684-90
9. Faje A. Hypophysitis: Evaluation and management. Clin Diabetes Endocrinol. 2016. 2: 1-8
10. Fedala NS, Chentli F, Meskine D, Haddam AE. Les hypophysites: Aspects cliniques et évolutifs. Ann Endocrinol. 2016. 77: 338
11. Folkerth RD, Price DL, Schwartz M, Black PM, De Girlami U. Xanthomatous hypophysitis. Am J Surg Pathol. 1998. 22: 736-41
12. Fukuoka H. Hypophysitis. Endocrinol Metab Clin North Am. 2015. 44: 143-9
13. Goudi RB, Pinkerton PH. Anterior hypophysitis and hashimoto’s disease in a young woman. J Pathol Bacteriol. 1962. 83: 584-5
14. Guo S, Wang C, Zhang J, Tian Y, Wu Q. Diagnosis and management of tumor-like hypophysitis: A retrospective case series. Oncol Lett. 2016. 11: 1315-20
15. Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Brück W, Caturegli P. Primary hypophysitis: Clinical-pathological correlations. Eur J Endocrinol. 2006. 155: 101-7
16. Gutenberg A, Larsen J, Lupi I, Rohde V, Caturegli P. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. AJNR Am J Neuroradiol. 2009. 30: 1766-72
17. Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C. Treatment of primary hypophysitis in Germany. J Clin Endocrinol Metab. 2015. 100: 3460-9
18. Honegger J, Fahlbusch R, Bornemann A, Hensen J, Buchfelder M, Müller M. Lymphocytic and granulomatous hypophysitis: Experience with nine cases. Neurosurgery. 1997. 40: 713-23
19. Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U. Diagnosis of primary hypophysitis in Germany. J Clin Endocrinol Metab. 2015. 100: 3841-9
20. Hunn BH, Martin WG, Simpson S, Mclean CA. Idiopathic granulomatous hypophysitis: A systematic review of 82 cases in the literature. Pituitary. 2014. 17: 357-65
21. Imber BS, Lee HS, Kunwar S, Blevins LS, Aghi MK. Hypophysitis: A single-center case series. Pituitary. 2015. 18: 630-41
22. Karaca Z, Kelestimur F. The management of hypophysitis. Minerva Endocrinol. 2016. 41: 390-9
23. Khare S, Jagtap VS, Budyal SR, Kasaliwal R, Kakade HR, Bukan A. Primary (autoimmune) hypophysitis: A single centre experience. Pituitary. 2015. 18: 16-22
24. Kristof RA, Van Roost D, Klingmüller D, Springer W, Schramm J. Lymphocytic hypophysitis: Non-invasive diagnosis and treatment by high dose methylprednisolone pulse therapy?. J Neurol Neurosurg Psychiatry. 1999. 67: 398-402
25. Lupi I, Manetti L, Raffaelli V, Lombardi M, Cosottini M, Iannelli A. Diagnosis and treatment of autoimmune hypophysitis: A short review. J Endocrinol Invest. 2011. 34: e245-52
26. Lupi I, Zhang J, Gutenberg A, Landek-Salgado M, Tzou SC, Mori S. From pituitary expansion to empty sella: Disease progression in a mouse model of autoimmune hypophysitis. Endocrinology. 2011. 152: 4190-8
27. Park SM, Bae JC, Joung JY, Cho YY, Kim TH, Jin SM. Clinical characteristics, management, and outcome of 22 cases of primary hypophysitis. Endocrinol Metab (Seoul). 2014. 29: 470-8
28. Roth C, Wilken B, Hanefeld F, Schröter W, Leonhardt U. Hyperphagia in children with craniopharyngioma is associated with hyperleptinemia and a failure in the downregulation of appetite. Eur J Endocrinol. 1998. 138: 89-91
29. Simmonds M. Über das vorkommen von riesenzellen in der hypophyse. Virch Arch Pathol Anat. 1917. 223: 281-90

