

- Department of Neurosurgery, Ehime University School of Medicine,
- Division of Diagnostic Pathology, Ehime University Hospital, Shitsukawa, Toon, Ehime, Japan.
Correspondence Address:
Akihiro Inoue
Department of Neurosurgery, Ehime University School of Medicine,
DOI:10.25259/SNI_544_2019
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Daisuke Kohno, Akihiro Inoue, Mana Fukushima, Tomoharu Aki, Shirabe Matsumoto, Satoshi Suehiro, Masahiro Nishikawa, Saya Ozaki, Seiji Shigekawa, Hideaki Watanabe, Riko Kitazawa, Takeharu Kunieda. Epithelioid glioblastoma presenting as multicentric glioma: A case report and review of the literature. 17-Jan-2020;11:8
How to cite this URL: Daisuke Kohno, Akihiro Inoue, Mana Fukushima, Tomoharu Aki, Shirabe Matsumoto, Satoshi Suehiro, Masahiro Nishikawa, Saya Ozaki, Seiji Shigekawa, Hideaki Watanabe, Riko Kitazawa, Takeharu Kunieda. Epithelioid glioblastoma presenting as multicentric glioma: A case report and review of the literature. 17-Jan-2020;11:8. Available from: https://surgicalneurologyint.com/surgicalint-articles/9844/
Date of Submission
25-Dec-2019
Date of Acceptance
31-Dec-2019
Date of Web Publication
17-Jan-2020
Abstract
Background: Epithelioid glioblastoma is a rare aggressive variant of glioblastoma multiforme (GBM), which was formally recognized by the World Health Organization classification of the central nervous system in 2016. Clinically, epithelioid GBMs are characterized by aggressive features, such as metastases and cerebrospinal fluid dissemination, and an extremely poor prognosis. A rare case of epithelioid GBM that was discovered as a multicentric glioma with different histopathology is reported.
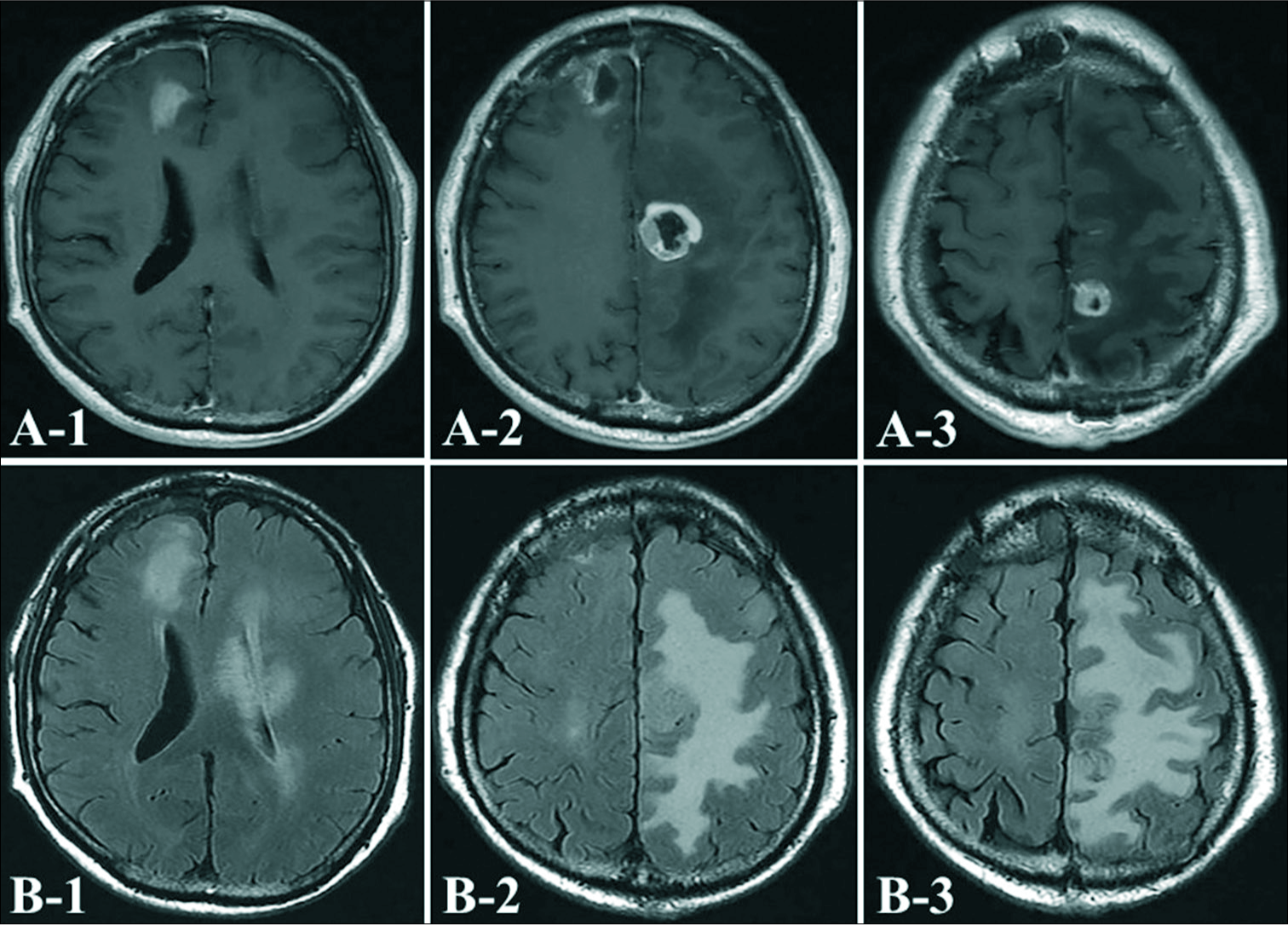
Case Description: A 78-year-old man was admitted to our hospital with mild motor weakness of the right leg. Neuroimaging showed small masses in the left frontal and parietal lobes on magnetic resonance imaging. The abnormal lesion had been increasing rapidly for 3 weeks, and a new lesion appeared in the frontal lobe. 11C-methionine positron emission tomography (PET) showed abnormal uptake corresponding to the lesion. To reach a definitive diagnosis, surgical excision of the right frontal mass lesion was performed. Histological findings showed diffuse astrocytoma. Only radiotherapy was planned, but the left frontal and parietal tumors progressed further within a short period. Therefore, it was thought that these tumors were GBM, and a biopsy of the left parietal tumor was performed. The histological diagnosis was epithelioid GBM. Immunohistochemistry showed that most tumor cells were negatively stained for p53 and isocitrate dehydrogenase 1. BRAF V600E mutations were not identified, but TERT promoter mutations were identified. Immediately after surgery, the patient was given chemotherapy using temozolomide, extended local radiotherapy and then bevacizumab. After 6 months, he showed no signs of recurrence.
Conclusion: Epithelioid GBM is one of the rarest morphologic subtypes of GBM and has a strongly infiltrative and aggressive nature. Therefore, careful identification of preoperative imaging studies and detailed evaluation of genetic studies are necessary to select the appropriate treatment for epithelioid GBM.
Keywords: Bevacizumab, BRAF V600E mutation, Epithelioid glioblastoma, Multicentric glioma, TERT promoter mutation
INTRODUCTION
Epithelioid glioblastoma multiforme (GBM) is one of the rarest aggressive GBM variants and was newly proposed in the World Health Organization (WHO) classification of the central nervous system (CNS) in 2016.[
CASE DESCRIPTION
A 78-year-old man presented to our department with mild motor weakness of the right leg. Magnetic resonance imaging (MRI) of the brain demonstrated small masses in the left frontal and parietal lobes [
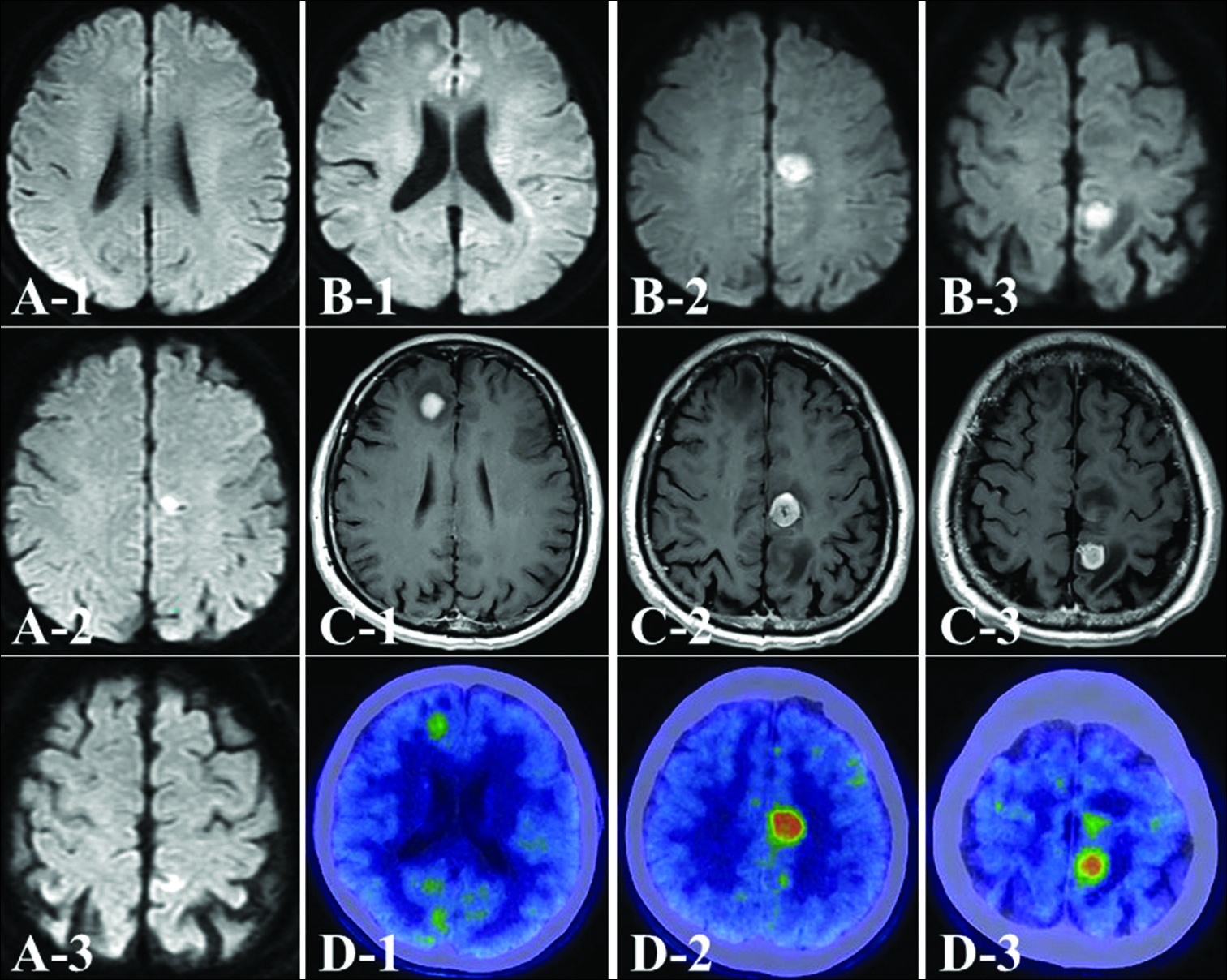
Figure 1:
Preoperative axial diffusion-weighted image (DWI) on magnetic resonance imaging shows a high-intensity mass in the left hemisphere (a). These masses are rapidly growing, and a new lesion appears in the right frontal lobe (b). These tumors in accordance with the DWI high-intensity lesion are homogeneously enhanced to a high degree with gadolinium (c). The maximum standardized uptake values on methionine positron emission tomography are high (d).
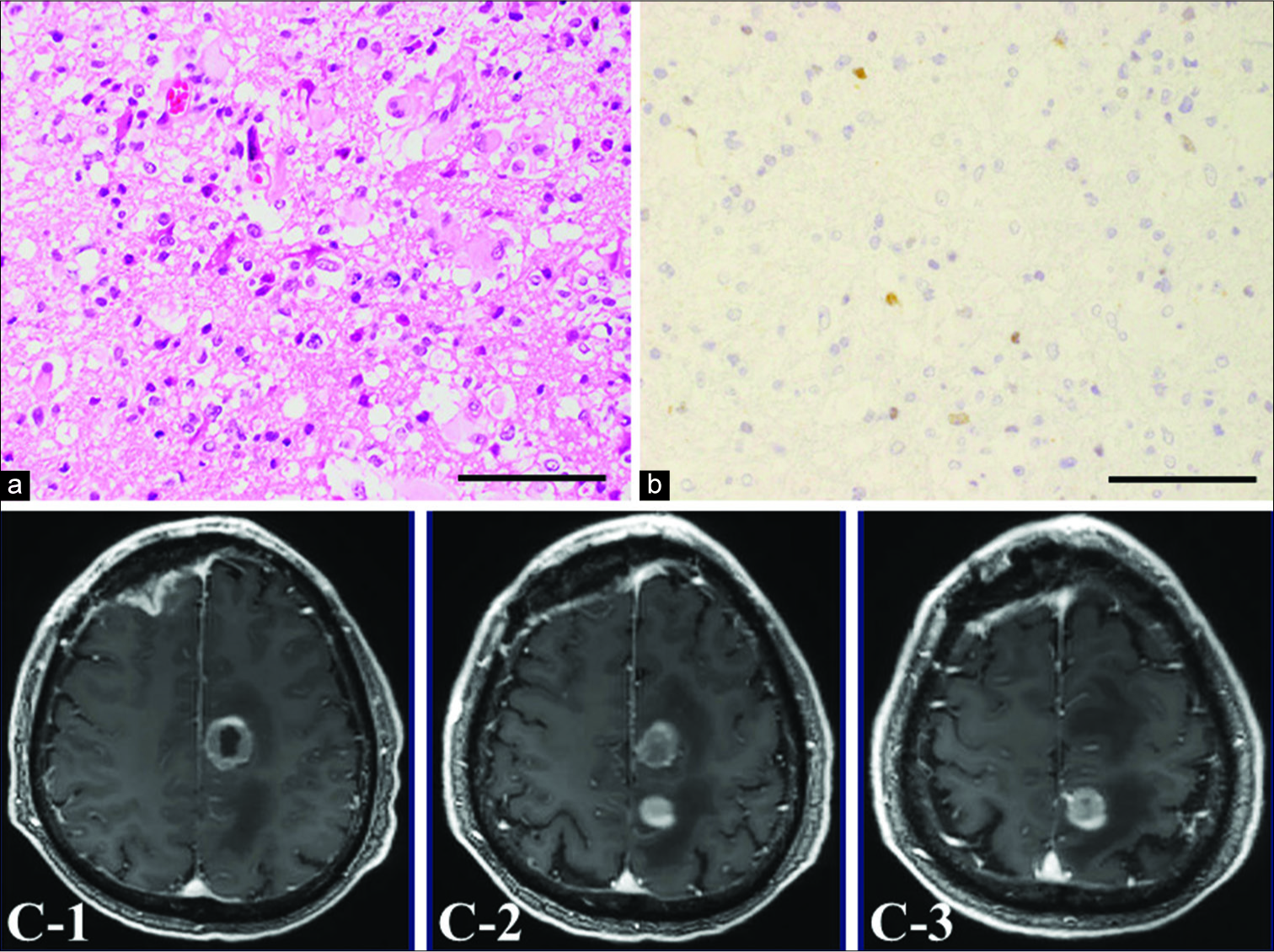
Figure 2:
Histopathology of the resected frontal tumor (a) shows diffuse astrocytoma. This tumor shows slightly positive staining for Ki-67 (b). Postoperative axial gadolinium-enhanced T1-weighted magnetic resonance imaging (c) demonstrates rapid growth within a short period. Magnification (a and b), ×400. Scale bar, 100 µm.
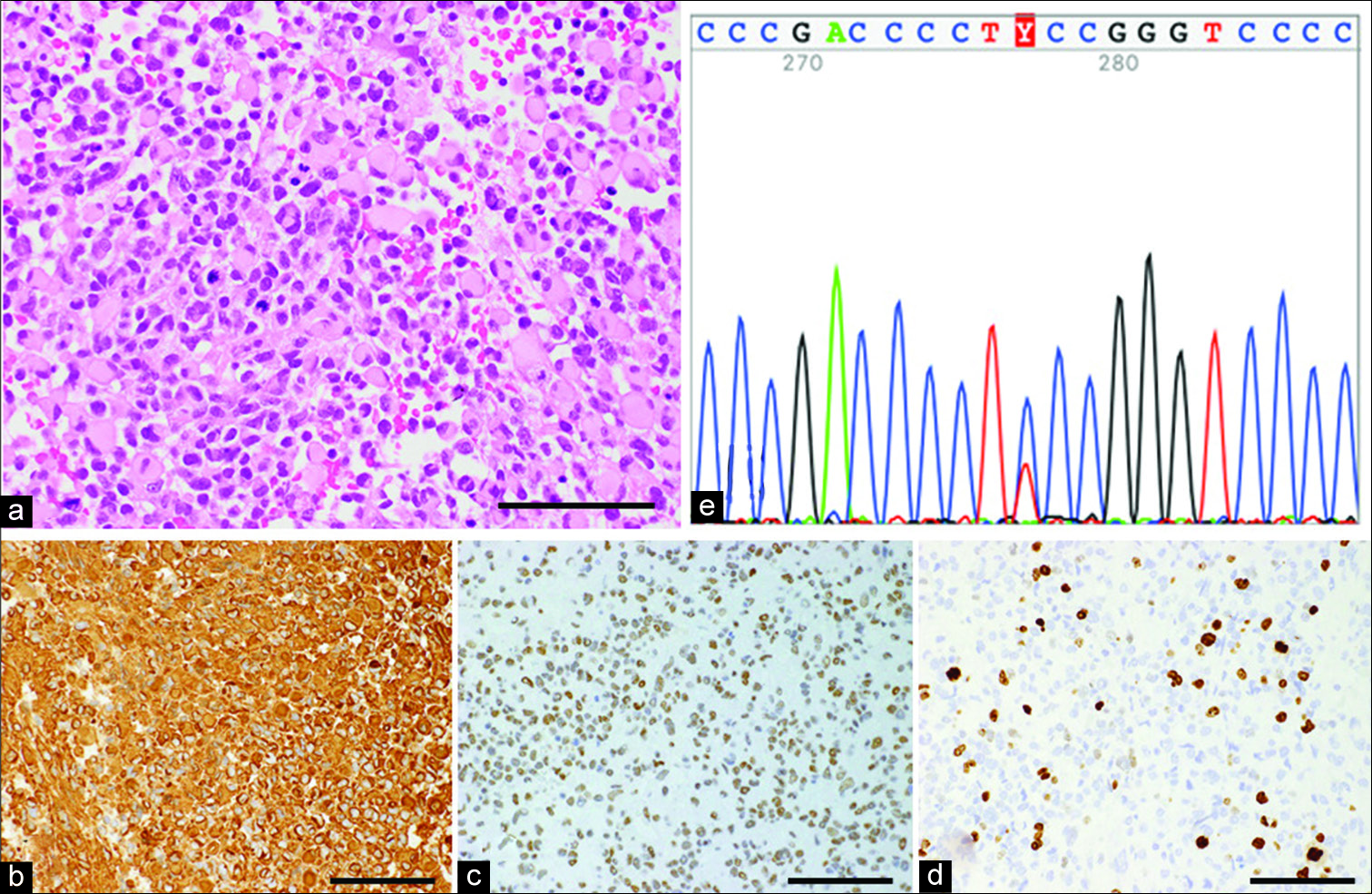
Figure 3:
Histopathology of the left parietal tumor. Hematoxylin and eosin staining (a) shows highly cellular and medium-sized rounded cells. Immunohistochemical examinations showing glial fibrillary acidic protein (b), integrase interactor 1 staining (c), and high Ki-67 labeling index (d). Magnification (a-d), ×400. Scale bar, 100 µm. Direct DNA sequence using Sanger method detects TERT promoter mutation (C250T: c.−146C>T) (e).
DISCUSSION
Epithelioid GBM is one of the rarest variants of GBM, and it was formally recognized in the 2016 WHO classification.[
In terms of imaging characteristics, preoperative identification of this tumor may be difficult with radiological examinations. In general, epithelioid GBM characteristically presents as a Gd-enhancing solid mass, occasionally with cysts. These tumors are prone to hemorrhage and often spread through the leptomeninges.[
A recent report based on integrated molecular analysis proposed that epithelioid GBM should be stratified into three subsets: a PXA subset, with a high percentage of BRAF V600E mutations, but a relatively low percentage of TERT promoter mutations; an adult IDH-wild-type GBM subset, with a relatively low percentage of BRAF V600E mutations, but a high percentage of TERT promoter mutations; and a pediatric RTK1 subset not harboring either mutation.[
With respect to the definition of multicentric gliomas, Batzdorf and Malamud reported criteria to distinguish multifocal tumors from multicentric tumors.[
Finally, with regard to the adjuvant therapy, the administration of bevacizumab was extremely effective in the present case. Recently, several case reports have described the success of targeted treatment using BRAF inhibitors and MEK inhibitors to BRAF V600E-mutant epithelioid GBM.[
CONCLUSION
Epithelioid GBM is a very rare, but well-documented entity. Epithelioid GBM should be considered in the differential diagnosis of multiple tumors due to its potential for highly aggressive and strong infiltrative features. Therefore, careful preoperative imaging with DWI/MRI and detailed evaluation of genetic studies including BRAF V600E and TERT promoter mutation are necessary for accurate diagnosis and appropriate selection of treatment for epithelioid GBM.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Batzdorf U, Malamud N. The problem of multicentric gliomas. J Neurosurg. 1963. 20: 122-36
2. Broniscer A, Tatevossian RG, Sabin ND, Klimo P, Dalton J, Lee R. Clinical, radiological, histological and molecular characteristics of paediatric epithelioid glioblastoma. Neuropathol Appl Neurobiol. 2014. 40: 327-36
3. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014. 370: 709-22
4. Cuncannon M, Wong M, Jayamanne D, Guo L, Cove N, Wheeler H. Role of delayed salvage bevacizumab at symptomatic progression of chemorefractory glioblastoma. BMC Cancer. 2019. 19: 445-
5. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014. 370: 699-708
6. He MX, Wang JJ. Rhabdoid glioblastoma: Case report and literature review. Neuropathology. 2011. 31: 421-6
7. Inoue A, Ohnishi T, Kohno S, Mizuno Y, Kitazawa R, Nakamura Y. A case of multicentric gliomas in both supra and infratentorial regions with different histology: A case report. World J Surg Oncol. 2016. 14: 152-
8. Kanemaru Y, Natsumeda M, Okada M, Saito R, Kobayashi D, Eda T. Dramatic response of BRAF V600E-mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: Establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol Commun. 2019. 7: 119-
9. Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol. 2013. 37: 685-98
10. Kleinschmidt-DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO. Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 but not claudin 6. Am J Surg Pathol. 2010. 34: 341-54
11. Korshunov A, Chavez L, Sharma T, Ryzhova M, Schrimpf D, Stichel D. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018. 28: 656-62
12. Kuroda J, Nobusawa S, Nakamura H, Yokoo H, Ueda R, Makino K. A case of an epithelioid glioblastoma with the BRAF V600E mutation colocalized with BRAF intact low-grade diffuse astrocytoma. Neuropathology. 2016. 36: 181-6
13. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
14. Malcolm FP, Thomas R, David GT. Multicentric malignant glioma. Br J Neurosurg. 1991. 5: 631-4
15. Mishra HB, Haran RP, Singh JP, Joseph T. Multicentric gliomas: Two case reports and a review of the literature. Br J Neurosurg. 1990. 4: 535-9
16. Momota H, Iwami K, Fujii M, Motomura K, Natsume A, Ogino J. Rhabdoid glioblastoma in a child: Case report and literature review. Brain Tumor Pathol. 2011. 28: 65-70
17. Nakajima N, Nobusawa S, Nakata S, Nakada M, Yamazaki T, Matsumura N. BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are frequent in epithelioid glioblastomas: A histological and molecular analysis focusing on intratumoral heterogeneity. Brain Pathol. 2018. 28: 663-73
18. Woo PY, Lam TC, Pu JK, Li LF, Leung RC, Ho JM. Regression of BRAFV600E mutant adult glioblastoma after primary combined BRAF-MEK inhibitor targeted therapy: A report of two cases. Oncotarget. 2019. 10: 3818-26

