

- Department of Oncology, University Hospital Southampton, Southampton, United Kingdom
- Department of Histopathology, University Hospital Southampton, Southampton, United Kingdom
Correspondence Address:
Georges Sinclair, Department of Oncology, University Hospital Southampton, Southampton, United Kingdom.
DOI:10.25259/SNI_190_2025
Copyright: © 2025 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Dinali Jayawardena1, Joseph Yates2, Enrico Clarke1, Georges Sinclair1. Extra-neural metastases of recurrent myxopapillary ependymoma: A patient case and literature review. 16-May-2025;16:182
How to cite this URL: Dinali Jayawardena1, Joseph Yates2, Enrico Clarke1, Georges Sinclair1. Extra-neural metastases of recurrent myxopapillary ependymoma: A patient case and literature review. 16-May-2025;16:182. Available from: https://surgicalneurologyint.com/surgicalint-articles/13559/
Date of Submission
21-Feb-2025
Date of Acceptance
14-Apr-2025
Date of Web Publication
16-May-2025
Abstract
Background: Biologically and morphologically distinct from other ependymomas, myxopapillary ependymomas (MPEs) are rare, slow-growing glial tumors originating predominantly from the conus medullaris, cauda equina, or filum terminale. Gross total resection is the standard of care for primary MPE. Nevertheless, despite maximal resection, the risk of recurrence, usually within the neural axis, remains high. However, extra-neural metastases can also occur. Due to the rarity of the entity, there is a lack of consensus on the management of recurrences and extra-neural metastatic disease. We present a case report and literature review of this rare ependymal tumor.
Case Description: We describe a case of a male patient with MPE who developed multiple recurrences, treated with numerous surgical resections, radiotherapy, and salvage chemotherapy before eventually developing extra-neural metastatic disease to lungs, abdomen, and lymph nodes 37 years after initial diagnosis. A biopsy of an axillary lymph node confirmed histomorphology comparable to the primary histology.
Conclusion: To our knowledge, there are
Keywords: Metastases, Myxopapillary ependymoma, Spine
INTRODUCTION
Biologically and morphologically distinct from other ependymomas,[
As such, MPE tends to recur, primarily locally, sometimes distally within the central nervous system (CNS) and rarely extra-neural.[
CASE DESCRIPTION
In 1986, a 24-year-old male presented to our hospital, UHS, with a year’s history of low back pain and right calf pain on exertion. The pain persisted despite a visit to the Chiropractor, an epidural injection, and analgesia. Shortly after, the patient developed right leg weakness with associated numbness in the soles of his feet; he also reported difficulty sustaining an erection, abnormal sensation on passing urine, and difficulty micturating. On emergency admission to the Spinal Unit, he had no relevant past medical history and was taking ibuprofen. A myelogram confirmed a complete block at T12, and cerebral spinal fluid (CSF) was slightly raised at 710 mg/L. An intramedullary lesion extending from T12 to the conus was suspected. He subsequently underwent a T12-L3 laminectomy and subtotal excision of the conus lesion at UHS. The ensuing histology showed papillae surrounded by myxoid material with strong diffuse staining for glial fibrillary acidic protein, findings consistent with a Myxopapillary Ependymoma World Health Organization (WHO) grade 1 (as per 2016 WHO classification and prior publications from 1979, 1993, 2000, and 2007) [
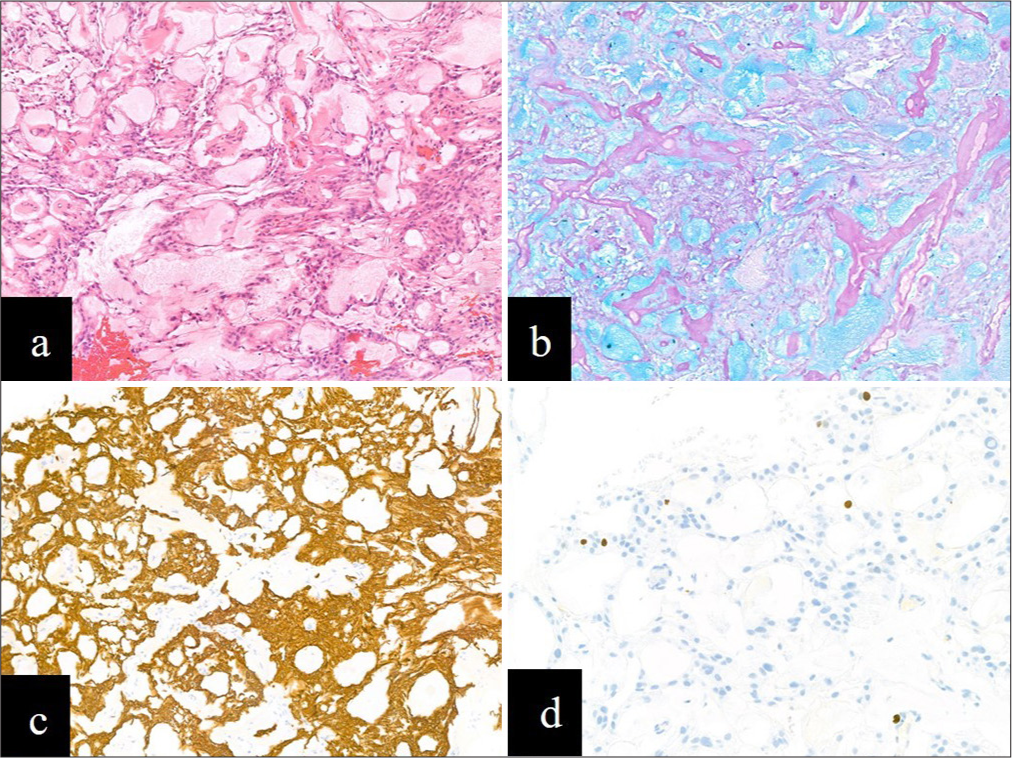
Figure 1:
(a) Haematoxylin and eosin section showing a tumor composed of papillae lined by a single layer of cuboidal cells and blood vessels with surrounding myxoid material (x100).(b) Alcian blue highlights the prominent mucin-rich collars around numerous blood vessels (x100). (c) The tumor cells showed strong and diffuse immunolabelling for glial fibrillary acidic protein (x100). (d) The Ki67 proliferation index was low at <3% (x200).
Approximately 20 months after his initial presentation (1988), he presented with a 3-week history of pain above his laminectomy site associated with paroxysmal, bilateral lower limb numbness. A repeat myelogram revealed a total block opposite T6. CSF cytology did not demonstrate malignant cells. A magnetic resonance imaging (MRI) scan showed a small focus of increased signal at the T7 level (images not available). Developing progressive symptoms, including a sensory level at T6, he underwent a second surgical intervention (thoracic laminectomy and total excision of recurrent T6 ependymoma at UHS). The corresponding histology examination revealed similar appearances to his initial biopsy, confirming recurrent MPE. Following surgery, he underwent craniospinal radiotherapy (35Gy in 21 fractions with a focal boost to the involved area of thoracic cord T5-T7, total dose 50Gy in 31 fractions at UHS). He made a complete recovery following surgery and radiotherapy, returning to work as a draughtsman and playing badminton.
He remained clinically and radiologically stable for the next 11 years before presenting with progressive numbness in his feet, extending up to buttocks, affecting mobility (1999). An MRI scan revealed a large tumor occupying the whole of the thecal sac from T12 to L5, with a second tumor at T10 causing cord compression (images not available). Consequently, the patient underwent emergency re-do thoracic laminectomy T9-T11 and lumbar laminectomy L3-L5 (third surgery at UHS); despite complete removal of the T10 lesion, the lumbar tumor could not be excised due to multiple nerve root infiltration. The subsequent histology was in keeping with previous examinations, i.e., recurrent MPE. He received further postoperative radiotherapy to T11-S2 (18 Gy in 10 fractions with a further boost below the conus of 5.4 Gy in 3 fractions at UHS). To consolidate treatment, the patient was offered postradiation chemotherapy with procarbazine, lomustine (CCNU), and vincristine 6-weekly (PCV chemotherapy). However, after 3 cycles, chemotherapy was discontinued due to concerns over bone marrow depletion and long bone marrow recovery. At this point, he had a Karnofsky performance status (KPS) of 90 with good power bilaterally but reduced sensation over thighs, buttocks, and perineal area with some difficulty in erectile function.
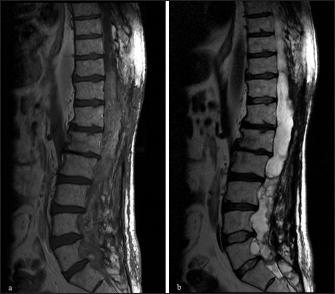
Approximately 13 years after his third surgery (2013), he had a rapid, progressive motor deterioration requiring a wheelchair. An MRI scan [

Approximately 10 months later (2014), an MRI scan [
Over the next 8 years, his tumor slowly progressed, resulting in worsening bowel and bladder function, leading to the insertion of a colostomy and suprapubic catheter. He remained reasonably well, still mobilizing on his wheelchair. However, in 2023, he presented to the GP with low oxygen saturations. A chest X-ray was arranged demonstrating a right pleural effusion; the complementary computed tomography (CT) of the chest, abdomen, and pelvis showed multiple bilateral lung nodules and enlarged nodes in the axillae, paraaortic, and retroperitoneal regions [
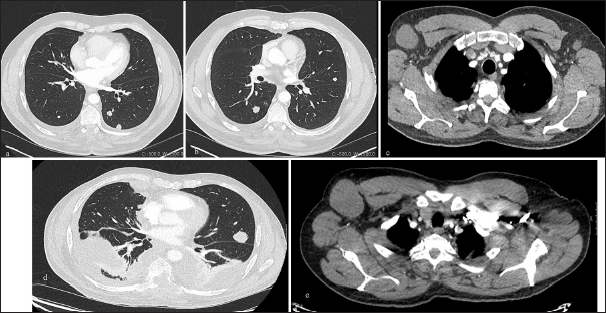
Figure 4:
Axial computed tomography (CT) slices at initial metastatic presentation demonstrating (a and b) metastatic bilateral pulmonary nodules and (c) enlarged axillary nodes. Axial CT slices 1 year later demonstrating (d) progression of disease with extensive thoracic metastatic disease, bilateral pleural effusions and (e) further enlargement of axillary nodes.
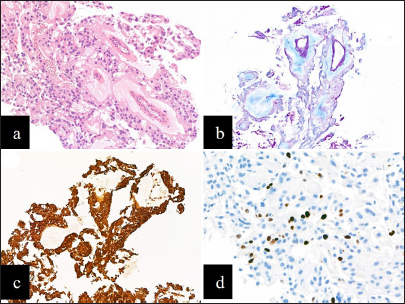
Figure 5:
(a) Haematoxylin and eosin section of the metastatic deposit of myxopapillary ependymoma showing similar histological features to the primary tumor (x100). (b) Alcian blue highlights abundant mucin around blood vessels (x100). (c) The tumor cells showed strong and diffuse expression of glial fibrillary acidic protein (x100). (d) The Ki67 proliferation index was low to focally moderate at 2.5-7.5% (x200).
His case was discussed at the National Ependymoma Multidisciplinary Group Meeting. The group concluded that treatment options were limited with no role of further radiotherapy. Oral etoposide was suggested, yet his clinical condition heavily precluded further chemotherapy. Consequently, the patient was referred to the palliative care team for best supportive management. Sadly, the patient passed away in late January 2025, 39 years after his initial diagnosis, at the age of 63. A chronological summary of the patient’s disease evolution and treatments is provided in
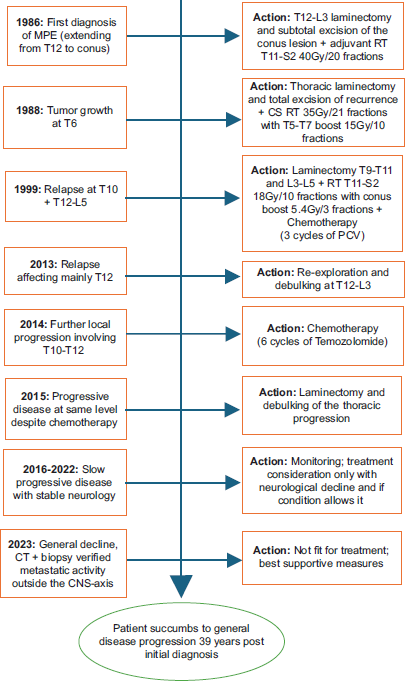
DISCUSSION
Background
Ependymomas are intramedullary tumors arising from ependymal glial cells lining the ventricles and central spinal canal.[
Although remaining an extremely rare occurrence, extra-neural metastases of MPEs have been reported in the lung, pleura, mediastinum, liver, bone, cervix, diaphragmatic, abdominal, pelvic muscles, and lymph nodes.[

In summary, extra-neural metastases of MPE are associated with worse prognosis;[
Treatment strategies-Surgery and radiotherapy
Obviously, due to the rarity of the entity, there is a lack of evidence on the best management of MPE at recurrence, particularly extra-neural metastatic disease. Consequently, most treatments are derived from previous case studies, series, or retrospective analytical work. The standard of care for initial MPE diagnosis is surgery, aiming to achieve a gross total resection (GTR).[
Indeed, the guidance for adjuvant radiotherapy varies depending on histological subtype and outcome at surgery.[
As mentioned previously, our patient survived almost four decades after the initial diagnosis despite experiencing several episodes of recurrence matching the timeline of survival of some of the cases identified by Mastantuoni et al.;[
Treatment strategies–Systemic treatment (chemotherapy, targeted therapy, and immunotherapy)
Often the case of multiple lines of local recurrence and/or distant metastases, further surgery and radiotherapy may not be feasible; in this framework, systemic therapies are contemplated and worth discussing. TMZ is often used in the treatment of glial tumors, with its use being extended to ependymal tumors. There is a level of evidence to suggest a response to TMZ in chemotherapy-naïve patients with a median PFS of 9.69 months and OS of 30.55 months of a retrospective study of 18 patients with recurrent intracranial ependymomas.[
Oral etoposide was suggested by the National Ependymoma Multidisciplinary Group Meeting as a treatment option for our patient. Unfortunately, he was unsuitable to start treatment. Yet cases with positive outcomes have been reported; a phase II trial comprising 10 patients with recurrent intramedullary spinal ependymomas was given oral etoposide, having failed surgery, radiotherapy, or salvage chemotherapy.[
Cisplatin has also been reported to have higher response rates compared with nonplatinum-based chemotherapy, but this did not translate to significant PFS or OS in a small retrospective analysis of 28 adults with recurrent intracranial ependymomas.[
Bevacizumab, a monoclonal antibody that binds to and inhibits vascular endothelial growth factor (VEGF), has been observed to have a PFS of 6.4 months and OS of 9.4 months in a retrospective case series of 8 patients with intracranial ependymomas treated with bevacizumab alone or in combination with other chemotherapy.[
Over the past 30 years, immunotherapy has shown remarkable benefits and is now frequently used as systemic therapy across multiple tumor sites; yet, its role is less well established in tumors of the CNS. Tapia Rico et al. successfully treated a patient with metastatic spinal MPE with the trial drug, tislelizumab, an anti-PD-1 antibody [
CONCLUSION
Although a rare occurrence, MPE can metastasize outside the CNS. To our knowledge, there are <30 cases of extra-cranial metastatic MPE reported since 1955, which heavily precludes the obtention of reliable clinical data and implementation of evidence-based guidelines. Consequently, there is no current consensus on the treatment of extra-neural metastatic MPE. With this in mind, case reports and series remain of utter importance to share experience and help customize management. From this angle, surgery, and radiotherapy are still used in the face of CNS recurrence and “limited” extra-neural spread, depending on the patterns of invasion, previous treatments, and KPS. Although chemotherapy’s efficiency on MPE remains questionable, positive outcomes from targeted agents and immunotherapy (alone or combined) have been reported, ultimately warranting further exploration. Finally, long-term, regular, follow-up of these patients is strongly recommended due to the long latency before metastatic presentation.
Ethical approval:
Institutional Review Board approval is not required.
Declaration of patient consent:
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship:
Fothergill Neuroscience Society (UK).
Conflicts of interest:
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation:
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
Acknowledgment:
Our sincere expression of gratitude to the Fothergill Neuroscience Society (UK) for kindly funding the publishing fees relevant to this case report, a much-appreciated gesture.
References
1. Akyurek S, Chang EL, Yu TK, Little D, Allen PK, McCutcheon I. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. Anderson cancer center. J Neurooncol. 2006. 80: 177-83
2. Almatrafi FR, Aseeri AM, Alqahtani MF, Al Mulla L, Al-Jubran S, AlOmair MA. Myxopapillary ependymoma. A case report of rare multicentric subtype and literature review. Med Arch. 2023. 77: 150-4
3. Al-Mistarehi AH, Parker M, Xia Y, Hasanzadeh A, Horowitz MA, Raj D. Survival factors in 1580 adults with spinal ependymoma: Insights from a multicenter oncology database. World Neurosurg. 2024. 190: e920-30
4. Batich KA, Riedel RF, Kirkpatrick JP, Tong BC, Eward WC, Tan CL. Recurrent extradural myxopapillary ependymoma with oligometastatic spread. Front Oncol. 2019. 9: 1322
5. Brandes AA, Cavallo G, Reni M, Tosoni A, Nicolardi L, Scopece L. A multicenter retrospective study of chemotherapy for recurrent intracranial ependymal tumors in adults by the gruppo italiano cooperativo di neuro-oncologia. Cancer. 2005. 104: 143-8
6. Celli P, Cervoni L, Cantore G. Ependymoma of the filum terminale: Treatment and prognostic factors in a series of 28 cases. Acta Neurochir (Wien). 1993. 124: 99-103
7. Chamberlain MC. Salvage chemotherapy for recurrent spinal cord ependymona. Cancer. 2002. 95: 997-1002
8. Deniel A, Marguet F, Beaussire L, Tobenas-Dujardin AC, Peillon C, Gambirasio MA. TERTp mutation detection in plasma by droplet-digital polymerase chain reaction in spinal myxopapillary ependymoma with lung metastases. World Neurosurg. 2019. 130: 405-9
9. Fecker A, Maanum KA, Shahin MN, Hakar M, Wright lii JM. Myxopapillary ependymoma metastasis mimicking pulmonary embolism: An illustrative case. Asian J Neurosurg. 2024. 19: 551-5
10. Fegerl G, Marosi C. Stabilization of metastatic myxopapillary ependymoma with sorafenib. Rare Tumors. 2012. 4: 134-7
11. Fujimori T, Iwasaki M, Nagamoto Y, Kashii M, Sakaura H, Yoshikawa H. Extraneural metastasis of ependymoma in the cauda equina. Global Spine J. 2013. 3: 33-40
12. Green RM, Cloughesy TF, Stupp R, DeAngelis LM, Woyshner EA, Ney DE. Bevacizumab for recurrent ependymoma. Neurology. 2009. 73: 1677-80
13. Guzin K, Bozdag H, Aydin A, Sahin S, Ozkanli S. Uterine cervix metastasis of myxopapillary ependymoma originated from the spinal cord. Balkan Med J. 2016. 33: 235-8
14. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, FigarellaBranger D. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021. 23: 1231-51
15. Mahalingam P, Smith S, Lopez J, Sharma RK, Millard T, Thway K. PARP inhibition utilized in combination therapy with olaparib-temozolomide to achieve disease stabilization in a rare case of BRCA1mutant, metastatic myxopapillary ependymoma. Rare Tumors. 2023. 15: 20363613231152333
16. Mastantuoni C, Tortora F, Tafuto R, Tortora M, Briganti F, Franca RA. Extra-neural metastases of late recurrent myxopapillary ependymoma to left lumbar paravertebral muscles: Case report and review of the literature. Brain Sci. 2022. 12: 1227
17. Oh MC, Ivan ME, Sun MZ, Kaur G, Safaee M, Kim JM. Adjuvant radiotherapy delays recurrence following subtotal resection of spinal cord ependymomas. Neuro Oncol. 2012. 15: 208-15
18. Riegel DC, Fonkem E, Connelly JM. Treatment of extraneural metastases of myxopapillary ependymomas with dose-dense temozolomide and lapatinib. Cureus. 2024. 16: e67928
19. Rudà R, Bosa C, Magistrello M, Franchino F, Pellerino A, Fiano V. Temozolomide as salvage treatment for recurrent intracranial ependymomas of the adult: A retrospective study. Neuro Oncol. 2015. 18: 261-8
20. Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, Santarius T. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018. 20: 445-56
21. Tapia Rico G, Townsend A, Price T, Patterson K. Metastatic myxopapillary ependymoma treated with immunotherapy achieving durable response. BMJ Case Rep. 2020. 13: e236242
22. Weber DC, Wang Y, Miller R, Villà S, Zaucha R, Pica A. Long-term outcome of patients with spinal myxopapillary ependymoma: Treatment results from the MD anderson cancer center and institutions from the rare cancer network. Neuro oncol. 2014. 17: 588-95
23. Welch WSchiff DGerszten P. Available from: https://www.uptodate.com/contents/spinal-cord-tumors?csi=34b9ae28-6109-43b3-8ec7-62f7b19617f1&source=contentshare [Last accessed on 2025 Jan 03].

