

- Department of Neurosurgery, “K.A.T.-N.R.C.” General Hospital of Attica,
- Department of Neurosurgery, 251 Hellenic Air Force General Hospital,
- Department of Neurosurgery, University of Athens, “Evangelismos Hospital”,
- Departments of Imaging, Children’s Hospital “Aghia Sofia”, Athens, Greece.
- Departments of Neurosurgery, Children’s Hospital “Aghia Sofia”, Athens, Greece.
Correspondence Address:
Dimitrios Giakoumettis
Departments of Neurosurgery, Children’s Hospital “Aghia Sofia”, Athens, Greece.
DOI:10.25259/SNI_10_2019
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Aikaterini Karagianni, Ploutarchos Karydakis, Dimitrios Giakoumettis, Ioannis Nikas, George Sfakianos, Marios Themistocleous. Fetal subependymal giant cell astrocytoma: A case report and review of the literature. 25-Feb-2020;11:26
How to cite this URL: Aikaterini Karagianni, Ploutarchos Karydakis, Dimitrios Giakoumettis, Ioannis Nikas, George Sfakianos, Marios Themistocleous. Fetal subependymal giant cell astrocytoma: A case report and review of the literature. 25-Feb-2020;11:26. Available from: https://surgicalneurologyint.com/surgicalint-articles/9878/
Date of Submission
08-Jan-2019
Date of Acceptance
04-Feb-2020
Date of Web Publication
25-Feb-2020
Abstract
Background:Subependymal giant cell astrocytomas (SEGAs) appear approximately in 10% of patients with tuberous sclerosis. These tumors are most commonly diagnosed in childhood and adolescence, with in utero diagnosed SEGAs being an extremely rare entity.
Case Description:We present the case of a congenital SEGA detected in an antenatal ultrasound and further investigated with fetal magnetic resonance imaging (MRI) scans at 22 and 32 weeks of gestational age. At 9 days of age, the child underwent craniotomy and partial excision of the tumor, followed by a second more extensive operation 13 days later. The patient was subsequently administered mammalian target of rapamycin inhibitor (everolimus).
Conclusion:In the latest follow-up MRI, at the age of two, the SEGA remained unchanged. Management of these tumors in neonates is challenging, mainly due to high morbidity and mortality of surgical treatment in these ages.
Keywords: Congenital, In utero diagnosis, Subependymal giant cell astrocytoma, Tuberous sclerosis
INTRODUCTION
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder of high penetrance. It is estimated that this condition affects 1 child in 6000[
CASE REPORT
Case illustration
We present the rare case of a congenital SEGA detected by fetal magnetic resonance imaging (MRI).
This female was the first child of healthy parents with no known family history of turner syndrome (TS). Abnormal findings in an antenatal ultrasound were further investigated with fetal MRI scans at 22 [
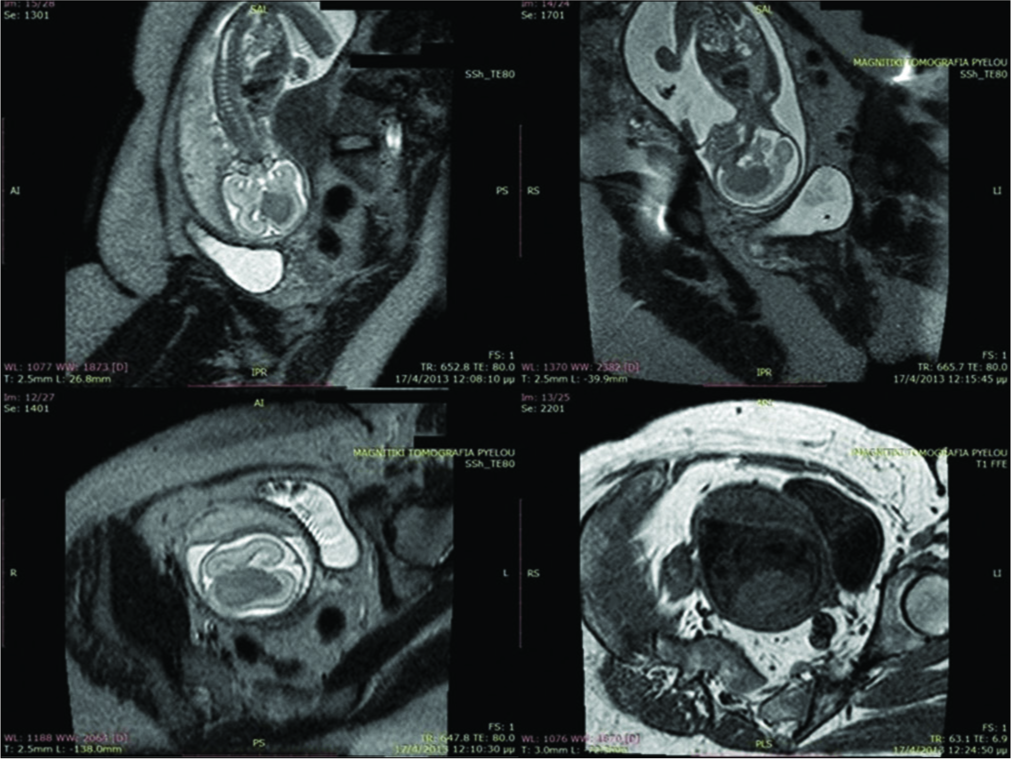
The girl was delivered by C-section at 38 weeks due to maternal hypertension. Apgar scores were 9 and 10 at 1 and 5 min, respectively, and the birth weight was 2690 g. Physical examination and routine hematology tests at admission were within normal values.
A postnatal ultrasound on day 2 showed a large lesion within the left lateral ventricle causing a midline shift to the right of 10 mm and mild dilatation of the third and right lateral ventricle. MRI on the same day [
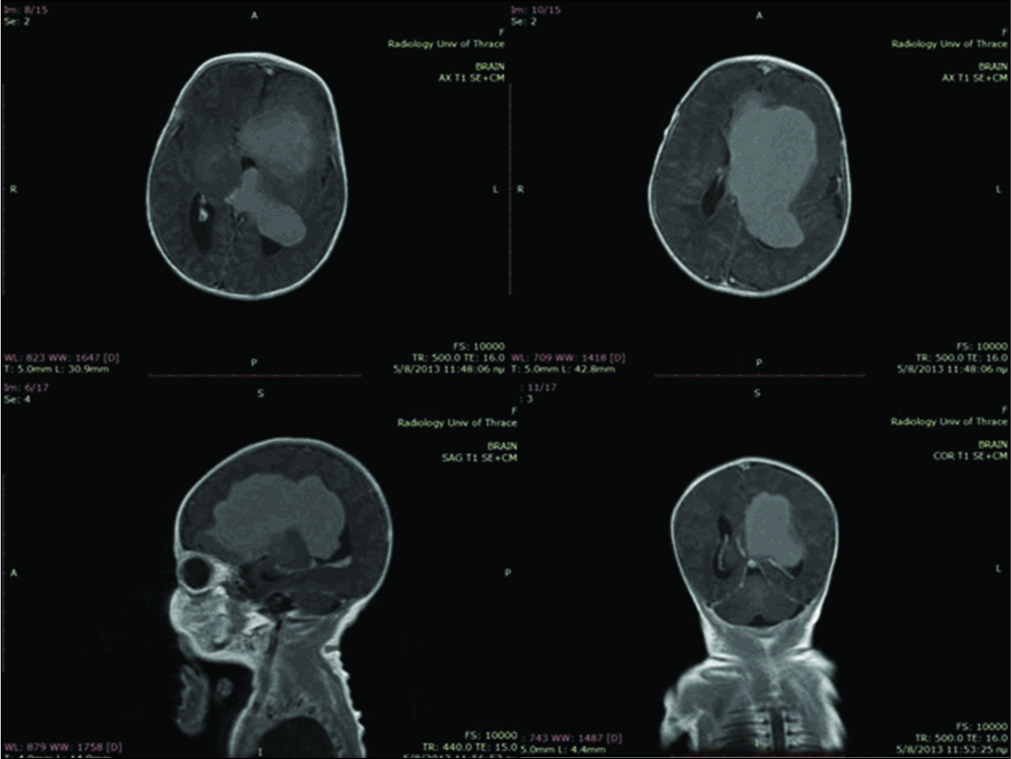
Figure 2:
Magnetic resonance imaging scans with gadolinium showing marked and rather homogeneous enhancement after gadolinium administration while part of the frontal lobe (straight gyrus) showed some peripheral enhancement indicative of local infiltration. A second smaller lesion of 4 mm was shown at the choroid plexus of the left lateral ventricle.
The patient was referred to the neurosurgical department of “Aghia Sofia” Children’s Hospital under the diagnosis of choroid plexus papilloma. At 9 days of age, the child underwent craniotomy and partial excision of the tumor, followed by a second, more extensive operation 13 days later.
Postoperatively, the patient was admitted to the intensive care unit both times. She was extubated after 3 days and a few hours, respectively. The postoperative course was uneventful and neurological examination of the patient remained unremarkable.
Histological examination of the material obtained, showed findings suggestive of a SEGA and concurrent presence of cortical tubers, therefore, establishing the diagnosis of tuberous sclerosis.[
Following the diagnosis of TS, the patient was subjected to additional investigations by echocardiogram, renal ultrasound, skin examination, ophthalmologic examination, and electroencephalography. No other manifestations of TS were found at that point.
During follow-up, seizures were noted for 2.5 months of age. An electroencephalogram showed the presence of single spike-wave.
A new effort to address the tumor by operation was performed at 4 months of age. Unfortunately, due to the infiltration of proximal structures (basal ganglia) and hemorrhagic nature of the lesion, total excision was not feasible. The patient was subsequently administered mammalian target of rapamycin (mTOR) inhibitor (everolimus). Two months later, during follow-up, a small angiomyolipoma of the left kidney was found.
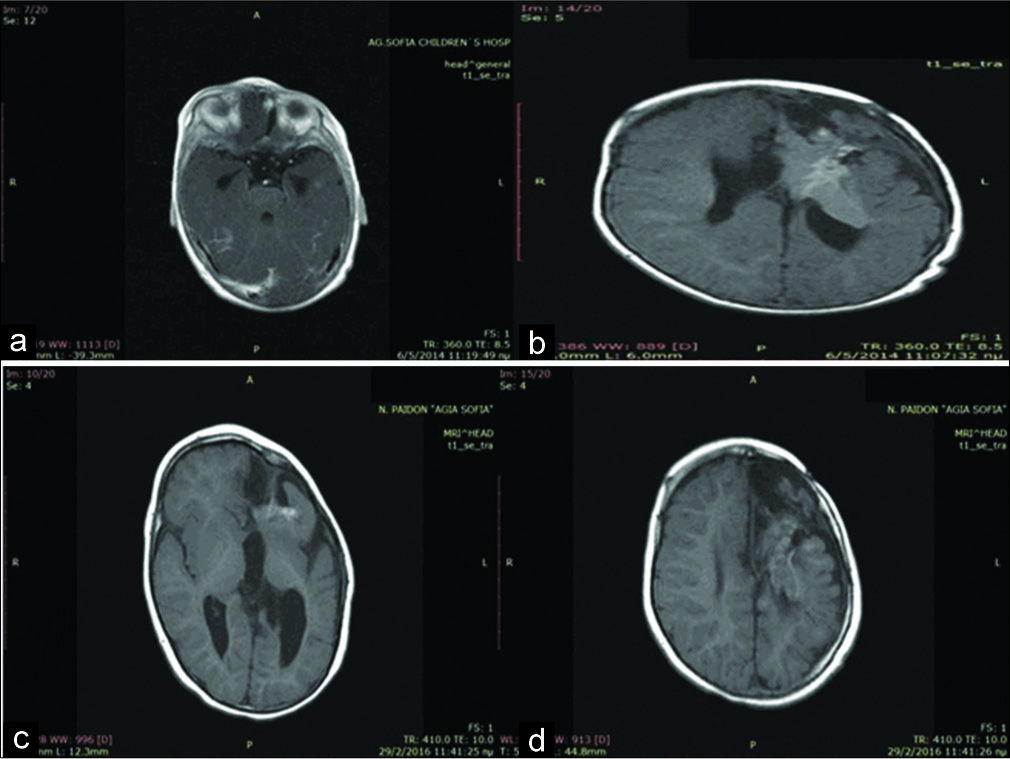
At 8 months of age, the patient was still receiving everolimus. Physical examination showed decreased mobility of the right hand. Atypical partial focal seizures appeared under treatment with vigabatrin, and the dose was modified. An MRI scan was obtained [
Pathology and immunohistochemical analysis
Pathological examination was carried out on hematoxylin and eosin stained paraffin-embedded tissue.
The tumor is composed of clusters of medium and large- sized polygonal and spindle-like eosinophilic cells. The nuclei of these cells contain prominent nucleoli. Their mitotic activity is very low. Among these cells, few gigantic neoplastic cells are noted with plump eosinophilic cytoplasm and either single or multiple nuclei without endonuclear vacuoles. Multiple psammoma bodies and calcium deposits are also present as well as a plethora of lymphocytes and macrophages [
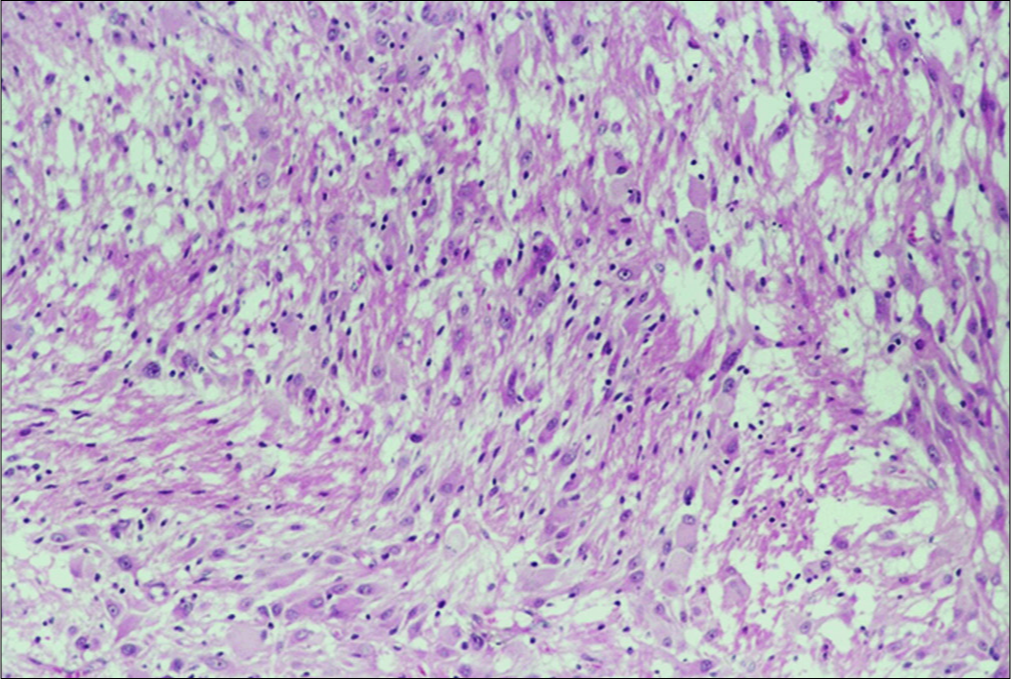
Within the brain parenchyma, areas of distorted architecture are also identified in the form of cortical tubers.
Medium-sized vessels (both arteries and veins) are identified within the tumor borders. They showed signs of wall tumor infiltration.
Immunofluorescence analysis showed that glial markers, glial fibrillary acidic protein [
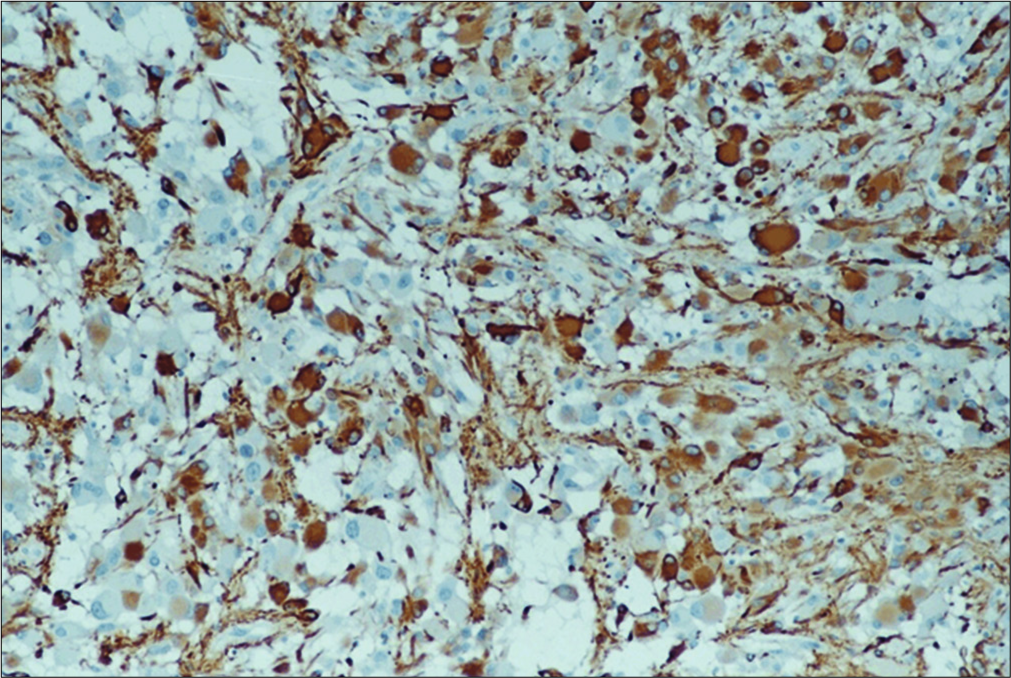
Ki-67/MIB-1 and p-53 were found positive only in 2–5% and 5% of the giant spindle-shaped cells, respectively, while bcl-2 was not detected.
Immunohistochemistry was positive for markers of T (CD3) differentiation of lymphocytes. Macrophages and microglial cells showed marked expression of CD68/PG- M1 markers.
DISCUSSION
Patients who present primarily with SEGA only rarely seem to manifest the complete classic triad of adenoma sebaceum, seizure, and mental retardation, which characterizes tuberous sclerosis. In neonates with SEGA, especially, the classic triad is highly unlikely to be found. In addition, the fact that a sporadic presentation is rather common with approximately 85% of the cases being the result of new mutations (therefore no familial history of TS is usually obtained), makes the preoperative diagnosis extremely challenging.
Regarding this specific case report, the preoperative diagnosis was choroid plexus papilloma due to the atypical radiological findings of the tumor, such as hyperintensity on T1-weighted MRI and hypointensity on T2-weighted MRI, in reference to the cerebral cortex. Such an atypical presentation has been reported in neonates as a result of higher water concentration in the neonate’s brain, calcification, and hypercellularity of the tumor.[
The tumor was located periventricularly, in proximity to the foramen of Monro, which is a characteristic location for SEGA.[
Following the establishment of the diagnosis, the choice of therapeutic approach seems to be equally challenging. Most patients with congenital SEGA undergo early surgery, although most reports document relatively poor surgical outcomes in neonates with a significantly high-risk operative morbidity and mortality.[
The reason lies in the fact that in neonates, the particular tumor seems to acquire more aggressive clinical features while the perioperative risk for major brain surgery in neonates is significantly high. The presence of cardiac rhabdomyomas also contributes, being the cause of cardiac complications and severe intraoperative arrhythmia.
On the contrary, massive intratumoral or intraventricular hemorrhage and acute hydrocephalus can cause rapid deterioration in these patients. Furthermore, tumor excision is believed to aid in decreasing the risk of developmental abnormalities elicited by TS. With the increasing use of mTOR inhibitor, a place for pharmaceutical treatment arises, perhaps combined with surgical procedure.[
Nonetheless, with thorough preoperative evaluation and meticulous planning of the treatment strategy, it is possible that the multiple issues resulting in the treatment of SEGA and its underlying condition in this fragile group of patients can be successfully addressed.
Thanks to the continuous technological advances made in the area of imaging, it is certain that more cases of congenital SEGA will be addressed in future, and the unique characteristics of the particular astrocytoma, as well as the ambiguous nature of tuberous sclerosis, are expected to be an increasing cause of controversy.
CONCLUSION
Even though SEGA is considered to be a rare entity, every mass in the proximity to the foramen of Monro should raise concern and suspicion to neurosurgeons for a subependymal giant cell astrocytoma in pediatric patients. Finally, comprehensive preoperative analysis and meticulous planning of the treatment strategy should be discussed in a multidisciplinary team of pediatric specialists, such as neurosurgeons, neurologists, neuroradiologists, pediatricians, and oncologists.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006. 355: 1345-56
2. Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004. 63: 1457-61
3. Hahn JS, Bejar R, Gladson CL. Neonatal subependymal giant cell astrocytoma associated with tuberous sclerosis: MRI, CT, and ultrasound correlation. Neurology. 1991. 41: 124-8
4. Hussain N, Curran A, Pilling D, Malluci CL, Ladusans EJ, Alfirevic Z. Congenital subependymal giant cell astrocytoma diagnosed on fetal MRI. Arch Dis Child. 2006. 91: 520-
5. Kotulska K, Borkowska J, Mandera M, Roszkowski M, Jurkiewicz E, Grajkowska W. Congenital subependymal giant cell astrocytomas in patients with tuberous sclerosis complex. Childs Nerv Syst. 2014. 30: 2037-42
6. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010. 363: 1801-11
7. Medhkour A, Traul D, Husain M. Neonatal subependymal giant cell astrocytoma. Pediatr Neurosurg. 2002. 36: 271-4
8. Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013. 49: 243-54
9. Oikawa S, Sakamoto K, Kobayashi N. A neonatal huge subependymal giant cell astrocytoma: Case report. Neurosurgery. 1994. 35: 748-50
10. Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991. 615: 125-7
11. Phi JH, Park SH, Chae JH, Hong KH, Park SS, Kang JH. Congenital subependymal giant cell astrocytoma: Clinical considerations and expression of radial glial cell markers in giant cells. Childs Nerv Syst. 2008. 24: 1499-503
12. Raju GP, Urion DK, Sahin M. Neonatal subependymal giant cell astrocytoma: New case and review of literature. Pediatr Neurol. 2007. 36: 128-31
13. Roth J, Roach ES, Bartels U, Jóźwiak S, Koenig MK, Weiner HL. Subependymal giant cell astrocytoma: Diagnosis, screening, and treatment. Recommendations from the international tuberous sclerosis complex consensus conference 2012. Pediatr Neurol. 2013. 49: 439-44
14. Tahiri Elousrouti L, Lamchahab M, Bougtoub N, Elfatemi H, Chbani L, Harmouch T. Subependymal giant cell astrocytoma (SEGA): A case report and review of the literature. J Med Case Rep. 2016. 10: 35-
15. Tien RD, Hesselink JR, Duberg A. Rare subependymal giant-cell astrocytoma in a neonate with tuberous sclerosis. AJNR Am J Neuroradiol. 1990. 11: 1251-2

