

Giant parieto-occipital lobe pediatric gliosarcoma: Report of a rare entity and review of literature
- Department of Neuro-Surgery, Govind Ballav Pant Institute of Postgraduate Medical Education and Research (GIPMER), New Delhi, India
Correspondence Address:
Gautam Dutta
Department of Neuro-Surgery, Govind Ballav Pant Institute of Postgraduate Medical Education and Research (GIPMER), New Delhi, India
DOI:10.4103/sni.sni_31_18
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Gautam Dutta, Robin Gupta, Manish Garg, Daljit Singh, Hukum Singh, Arvind K. Srivastava, Anita Jagetia. Giant parieto-occipital lobe pediatric gliosarcoma: Report of a rare entity and review of literature. 29-May-2018;9:111
How to cite this URL: Gautam Dutta, Robin Gupta, Manish Garg, Daljit Singh, Hukum Singh, Arvind K. Srivastava, Anita Jagetia. Giant parieto-occipital lobe pediatric gliosarcoma: Report of a rare entity and review of literature. 29-May-2018;9:111. Available from: http://surgicalneurologyint.com/surgicalint-articles/giant-parieto%e2%80%91occipital-lobe-pediatric-gliosarcoma-report-of-a-rare-entity-and-review-of-literature/
Date of Submission
27-Jan-2018
Date of Acceptance
23-Apr-2018
Date of Web Publication
29-May-2018
Abstract
Background:Gliosarcoma is a rare high-grade malignant tumor and a variant of glioblastoma characterized by biphasic glial and mesenchymal components. Gliosarcomas occur most commonly in the fifth or sixth decade of life and have a temporal lobe predilection. Occurrence in the pediatric population is extremely rare.
Case Description:Here, we report the case of an 8-year-old child with histologically confirmed gliosarcoma at the parieto-occipital lobe. Only a subtotal resection of the tumor mass could be performed in view of massive bleeding from the tumor bed; and despite postoperative chemotherapy and radiotherapy, the tumor recurred in a short span of time. A repeat surgery was done but the patient could not survive.
Conclusion:To our knowledge, this case constitutes the second youngest case reported in the literature with the lesion in the parieto-occipital region and the third youngest in all pediatric cases of gliosarcoma. This case demonstrates that possibility of gliosarcoma should always be kept in mind in children presenting with features of intracranial high-grade glial tumor. This case also suggests that significant residual after surgery is one variable that may affect the prognosis despite radiotherapy and/or chemotherapy.
Keywords: Brain neoplasm, glioblastoma multiforme, gliosarcoma, pediatric brain tumor
INTRODUCTION
Gliosarcoma (GS), a rare high-grade malignant tumor, is a variant of glioblastoma (GBM) characterized by biphasic glial and mesenchymal components.[
CASE REPORT
An 8-year-old child was referred to our hospital for evaluation and management of a mass lesion at the left parieto-occipital region. The patient gave a history of generalized tonic–clonic seizure episodes for the past 2 months along with holocranial headache and intermittent vomiting. There was no history of any weakness of any side of the body. On examination, the patient was lethargic but conscious, oriented, and cooperative. Neurological examination was normal. Contrast-enhanced computed tomography (CECT) of the brain was suggestive of a contrast enhancing mass in the left parieto-occipital region having a solid cystic component [
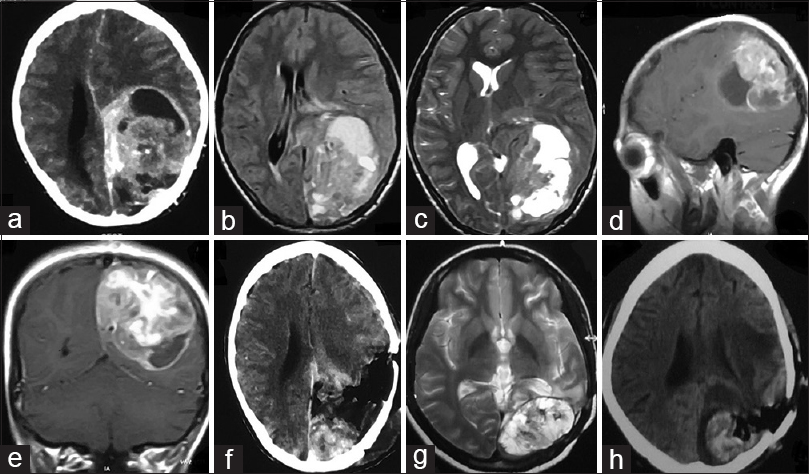
Figure 1
(a) CECT brain showing solid-cystic contrast enhancing mass left parieto-occipital lobe. (b) T1W MRI brain and (c) T2W MRI brain showing heterogenous signal intensity. (d) Sagittal and (e) coronal cuts of contrast MRI brain showing avid contrast enhancement. (f) CECT brain obtained after surgical resection of the mass showing significant residual tumor. (g) T2W MRI brain done after 6 months of surgery showing recurrent tumor. (h) Postoperative CT brain showing residual tumor and tumoral bed hematoma
The patient underwent left parieto-occipital craniotomy and decompression of the mass. The tumor was found to have both solid and cystic component with the cystic part containing xanthochromic fluid. The solid part was grayish and firm in consistency and highly vascular with ill-defined plane of cleavage. Subtotal removal was performed in view of severe blood loss [
Histology revealed highly vascular spindle cell tumor which was present in the form of sheets and fascicles. The cells had enlarged round to elongated nuclei, predominant nucleoli, and eosinophilic cytoplasm. Numerous mitotic figures, areas of necrosis, microvascular proliferation, and bizarre giant cells were seen. The cells were reticulin-rich and showed diffuse vimentin positivity and focal scattered glial fibrillary acidic protein (GFAP) positivity [Figure
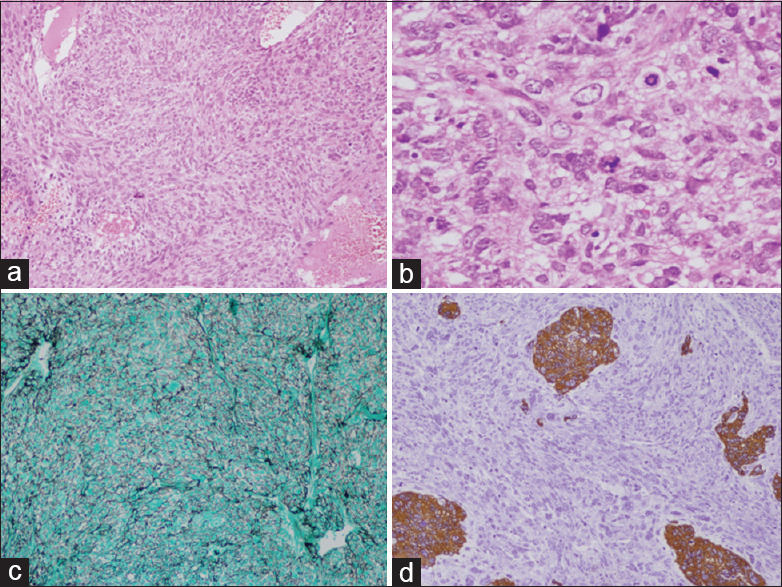
Figure 2
(a) H and E ×10 Gliosarcoma spindle cells in short fascicles with fibrillar background. (b) H and E ×40 showing numerous atypical mitosis. (c) Reticulin stain GMS ×10 showing numerous reticulin black fibers surrounding tumor cells. (d) Immunohistochemistry ×10 GFAP stain showing few islands of tumor cells as brown staining, rest sarcomatous components are stainless
Postoperative stay was uneventful and the patient was discharged on day 8. There was no neurological deficit and the patient improved symptomatically. As GS is known to spread extraneurally, CT abdomen and chest and MRI of the whole spine was done, which was negative. Concurrent chemoradiation of 60 Gy along with temozolomide 75 mg/m2 on all days of radiation was given. However due to financial constraints, patient stopped taking temozolomide further.
Six months after the surgery, the patient returned with disoriented behavior, headache, generalized tonic–clonic seizure episodes, and bouts of vomiting. A repeat MRI was obtained which was suggestive of recurrent lesion [
DISCUSSION
GS, defined as a variant of isocitrate dehydrogenase (IDH) wild-type glioblastoma, comprise 1.8–2.8% of glioblastomas and are rare biphasic tumors of the central nervous system, composing of alternating areas of glioblastomatous component admixed with sarcomatous component.[
GS is characterized by alternating areas of glial and mesenchymal differentiation – a gliomatous component – which expresses GFAP and is reticulin free, and a sarcomatous component, which lacks GFAP expression and is reticulin rich. The recent theory of pathogenesis of GS suggests monoclonal origin of both components of GS with sarcomatous component originating via aberrant mesenchymal differentiation of the malignant glioma. This theory explains the absence of significant difference in the clinical outcome between GBM and GS.[
GS and GBM are considered to be two different pathologies with some studies showing a temporal lobe predilection for GS whereas others found no such difference.[
The presenting signs and symptoms are consistent with those of rapidly expanding space occupying lesion such as headache and vomiting along with hemiparesis, seizures, and cognitive decline,[
On CECT scans, the lesions can appear with large necrotic areas and showing heterogeneous contrast enhancement, similar to GBM or as a hyperdense lesion with well-defined margins and showing homogenous enhancement, similar to that of meningioma.[
The histologic features include fascicles of sarcomatous component, usually resembling a fibrosarcoma or malignant fibrous histiocytoma, interspersed with areas of typical glioblastomatous component, thus creating a biphasic arrangement. Some cases may show a distinctive epithelial histology showing squamoid or glandular appearances which are immunonegative for GFAP, thus creating not only diagnostic dilemmas but also management difficulties for the neurosurgeons regarding whether these areas represent metastasis or a primary manifestation of a high-grade glial neoplasm.[
Treatment options for GS include maximum safe tumor resection followed by postoperative radiotherapy and chemotherapy.[
Role of temozolomide (TMZ) as an adjuvant therapy in GS remains controversial. Two earlier studies found no significant survival benefit with TMZ[
As with any tumors, total resection is of paramount importance for recurrence-free survival. However, in case of GS, this may not always be feasible as evident from the current case due to the high vascular nature of the tumor. Our patient had significant residual, and although postoperative RT and TMZ was administered, had recurrence in a short span of time; despite a repeat surgery, the patient could not survive. This may be a significant shortcoming of the treatment.
CONCLUSION
Although primarily a disease of the adult population, GS can rarely occur in pediatric group and this possibility should always be kept in mind in dealing with intracranial space occupying lesion in a child. Diagnosis and management can be clinically challenging with disheartening postoperative outcome due to its highly malignant behavior. More studies are needed to decide the best management option for childhood GS. Gross total resection should be attempted followed by postoperative radiotherapy and TMZ which may enhance patient outcome.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Actor B, Ludwig Cobbers JMJ, Büschges R, Wolter M, Knobbe CB, Lichter P. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer. 2002. 34: 416-27
2. Adeberg S, Bernhardt D, Harrabi SB, Diehl C, Koelsche C, Rieken S. Radiotherapy plus concomitant temozolomide in primary gliosarcoma. J Neurooncol. 2016. 128: 341-8
3. Castelli J, Feuvret L, Haoming QC, Biau J, Jouglar E, Berger A. Prognostic and therapeutic factors of gliosarcoma from a multi-institutional series. J Neurooncol. 2016. 129: 85-92
4. Feigin I, Allen LB, Lipkin L, Gross SW. The endothelial hyperplasia of the cerebral blood vessels with brain tumors, and its sarcomatous transformation. Cancer. 1958. 11: 264-77
5. Han SJ, Yang I, Ahn BJ, Otero JJ, Tihan T, McDermott MW. Clinical characteristics and outcomes for a modern series of primary gliosarcoma patients. Cancer. 2010. 116: 1358-66
6. Han SJ, Yang I, Tihan T, Prados MD, Parsa AT. Primary gliosarcoma: Key clinical and pathologic distinctions from glioblastoma with implications as a unique oncologic entity. J Neurooncol. 2010. 96: 313-20
7. Lee D, Kang SY, Suh YL, Jeong JY, Lee JI, Nam DH. Clinicopathologic and genomic features of gliosarcomas. J Neurooncol. 2012. 107: 643-50
8. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK.editors. Gliosarcoma. WHO classification of tumours of the central nervous system. Lyon: IARC; 2007. p. 48-9
9. Lutterbach J, Guttenberger R, Pagenstecher A. Gliosarcoma: A clinical study. Radiother Oncol. 2001. 61: 57-64
10. Nagaishi M, Paulus W, Brokinkel B, Vital A, Tanaka Y, Nakazato Y. Transcriptional factors for epithelial differentiation in gliosarcoma. Brain Pathol. 2012. 22: 670-6
11. Ozolek JA, Finkelstein SD, Couce ME. Gliosarcoma with epithelial differentiation: Immunohistochemical and molecular characterization. A case report and review of the literature. Mod Pathol. 2004. 17: 739-45
12. Parekh HC, O'Donovan DG, Sharma RR, Keogh AJ. Primary cerebral gliosarcoma: Report of 17 cases. Br J Neurosurg. 1995. 9: 171-8
13. Rath G, Sharma D, Mallick S, Gandhi AK, Joshi NP, Haresh KP. Clinical outcome of patients with primary gliosarcoma treated with concomitant and adjuvant temozolomide: A single institutional analysis of 27 cases. Indian J Cancer. 2015. 52: 599-
14. Ravisankar S, Chander RV, Devadoss PK. Pediatric gliosarcoma with fibrosarcomatous differentiation: Report of a rare case. Indian J Pathol Microbiol. 2012. 55: 521-4
15. Reis RM, Könü-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H. Genetic profile of gliosarcomas. Am J Pathol. 2000. 156: 425-32
16. Sampaio L, Linhares P, Fonseca J. Detailed magnetic resonance imaging features of a case series of primary gliosarcoma. Neuroradiol J. 2017. 30: 546-53
17. Savant HV, Balasubramaniam S, Mahajan V. Giant parietal lobe infantile gliosarcoma in a 5-year-old child. J Pediatr Neurosci. 2015. 10: 159-61
18. Walker C, Joyce KA, Thompson-Hehir J, Davies MP, Gibbs FE, Halliwell N. Characterisation of molecular alterations in microdissected archival gliomas. Acta Neuropathol. 2001. 101: 321-33
19. Walker GV, Gilbert MR, Prabhu SS, Brown PD, McAleer MF. Temozolomide use in adult patients with gliosarcoma: An evolving clinical practice. J Neurooncol. 2013. 112: 83-9
20. Zhang BY, Chen H, Geng DY, Yin B, Li YX, Zhong P. Computed tomography and magnetic resonance features of gliosarcoma: A study of 54 cases. J Comput Assist Tomogr. 2011. 35: 667-73

