

- Department of Neurosurgery, Institute of Medicine, University of Tsukuba, Ibaraki, Tsukuba, Japan.
- Department of Pediatrics, Institute of Medicine, University of Tsukuba, Ibaraki, Tsukuba, Japan.
- Department of Diagnostic Pathology, Institute of Medicine, University of Tsukuba, Ibaraki, Tsukuba, Japan.
Correspondence Address:
Ai Muroi, Department of Neurosurgery, Institute of Medicine, University of Tsukuba, Ibaraki, Tsukuba, Japan.
DOI:10.25259/SNI_405_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Erika Yamada1, Ai Muroi1, Ryoko Suzuki2, Hiroyoshi Kino1, Noriaki Sakamoto3, Takao Tsurubuchi1, Eiichi Ishikawa1. Infant-type hemispheric glioma occurring at the cervicomedullary region in a 5-month-old infant: A case report with a special emphasis on molecular classification. 25-Aug-2023;14:299
How to cite this URL: Erika Yamada1, Ai Muroi1, Ryoko Suzuki2, Hiroyoshi Kino1, Noriaki Sakamoto3, Takao Tsurubuchi1, Eiichi Ishikawa1. Infant-type hemispheric glioma occurring at the cervicomedullary region in a 5-month-old infant: A case report with a special emphasis on molecular classification. 25-Aug-2023;14:299. Available from: https://surgicalneurologyint.com/surgicalint-articles/12514/
Date of Submission
09-May-2023
Date of Acceptance
07-Aug-2023
Date of Web Publication
25-Aug-2023
Abstract
Background: High-grade gliomas in infancy are uncommon and have different clinical and molecular characteristics from those in adults. Recently, advances in molecular diagnostics have made progress in determining treatment strategies; however, the robust treatment has not yet been elucidated. We, herein, present a case of infantile glioma occurring at the cervicomedullary region.
Case Description: A 5-month-old infant developed left upper limb weakness and torticollis at 3 months of age. Magnetic resonance imaging revealed T2 hyperintensity from the medulla oblongata to the upper cervical cord. She underwent a biopsy for the lesion and pathological examination findings confirmed the presence of a high-grade astrocytoma with IDH wildtype-, H3K27M wildtype-, BRAF wildtype-, and ETV-NTRK3 fusion-positivity. Postoperatively, she underwent chemoradiotherapy, but she had marked tumor growth during the treatment. According to the new World Health Organization classification, the patient’s tumor is an infantile “hemispheric” glioma.
Conclusion: The characteristics and prognosis of NTRK-fused glioma are not fully understood, it is noteworthy that these tumors commonly occur in the brainstem. Further studies are needed to determine the prognosis of each tumor type and its sensitivity to treatment. This information will help in the reclassification of the tumors and identification of the precise treatment of this rare type of tumor.
Keywords: Astrocytic tumor, Cervicomedullary tumor, Glioma, Infant, NTRK
INTRODUCTION
High-grade gliomas (HGGs) in infants are rare, and the prevalence of astrocytoma and anaplastic astrocytoma in patients aged less than a year in Japan is reported to be 0.9% and 0.5%, respectively.[
CASE ILLUSTRATION
A 5-month-old infant had no particular abnormalities at birth or any progressing disease. At 3 months of age, she was brought to a regional clinic due to poor left upper limb movement. Brain magnetic resonance imaging (MRI) demonstrated a brainstem tumor; thus, she was referred to our hospital.
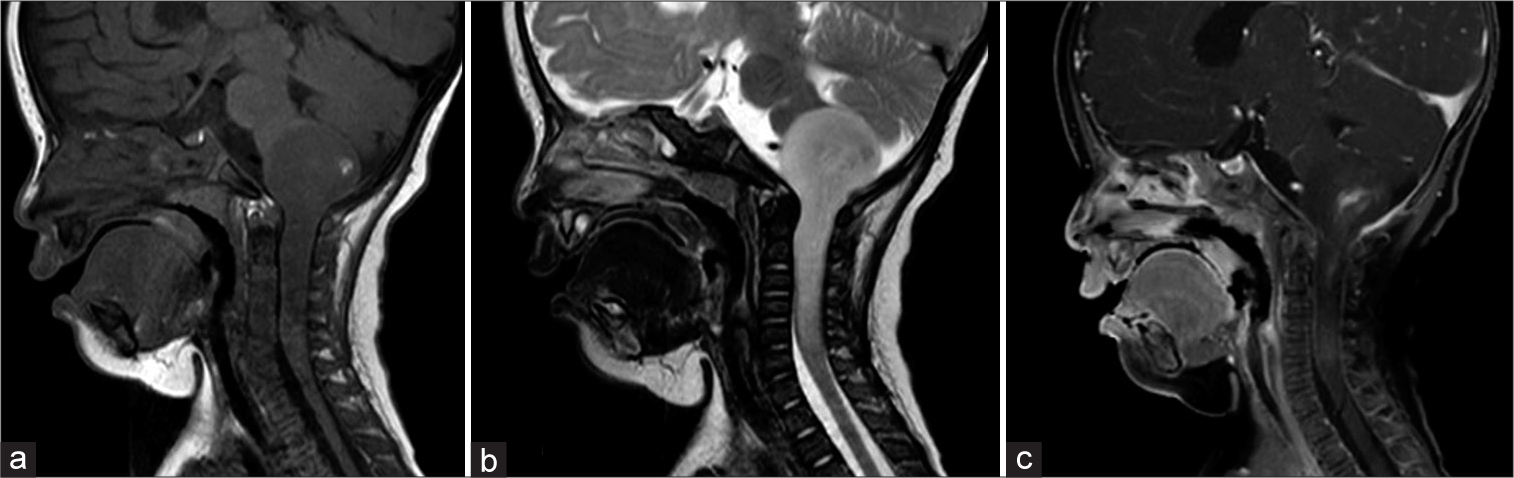
At the first visit at 5 months of age, she had right torticollis and left upper limb weakness. Both anterior and posterior fontanelles were closed. Her height, weight, and head circumference were 65.8 cm (+0.5 standard deviation [SD]), 6.3 kg (−0.9 SD), and 41 cm (−0.2 SD), respectively. Brain MRI showed an intrinsic mass extending from the medulla oblongata to the upper cervical cord. The tumor appeared uniformly hypointense on the T1-weighted image and hyperintense on the T2-weighted image with heterogeneous enhancement [
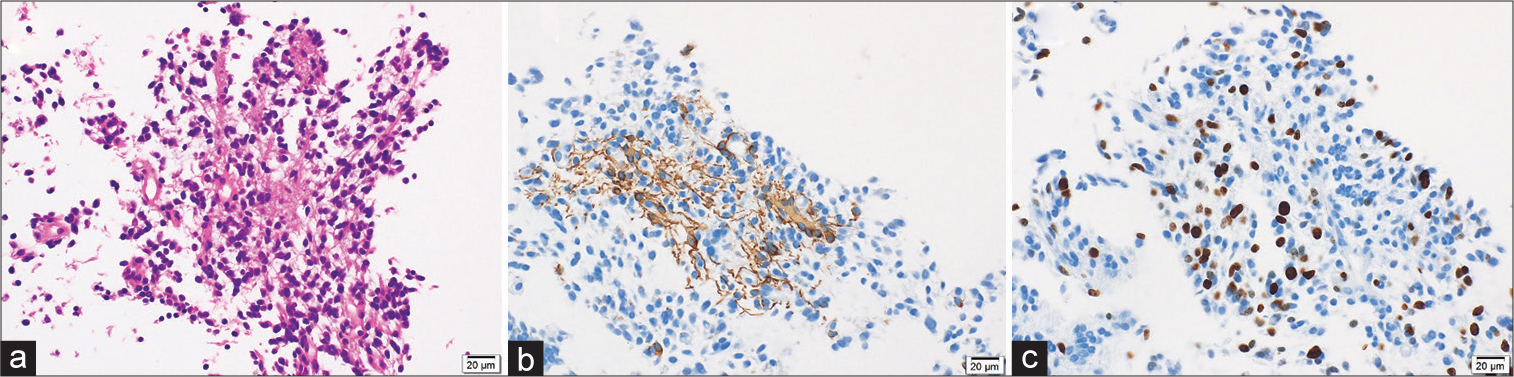
Histopathological findings included many atypical figures of nuclear fusion despite the absence of microvascular proliferation or necrosis. Small cells proliferated densely, nuclei were unevenly distributed, and eosinophilic protrusions were observed [
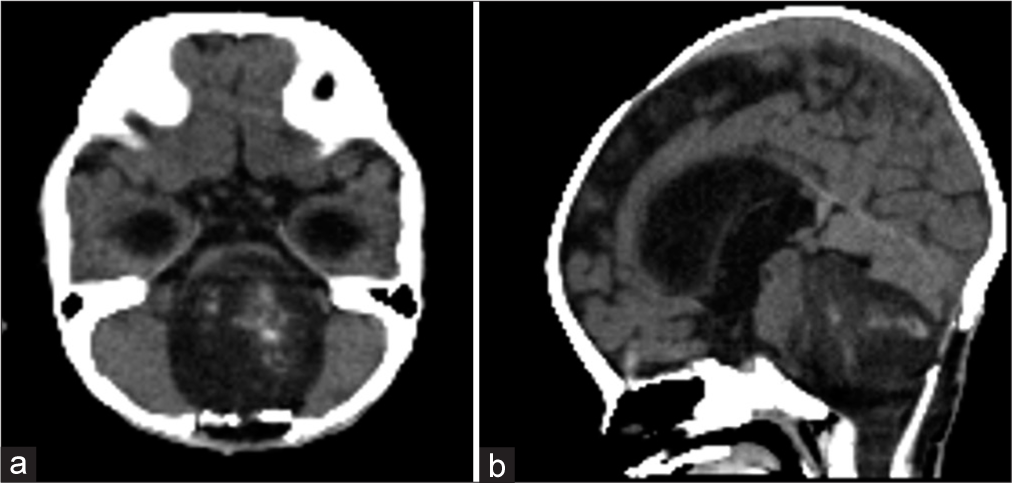
Based on the histological diagnosis, chemotherapy with baby Pediatric Oncology Group (POG) protocol (POG Trial 9233 Protocol) was initiated 3 weeks after the biopsy. However, the left-sided hemiparesis progressed on the 1st day of chemotherapy, and computed tomography showed an increase in tumor size [
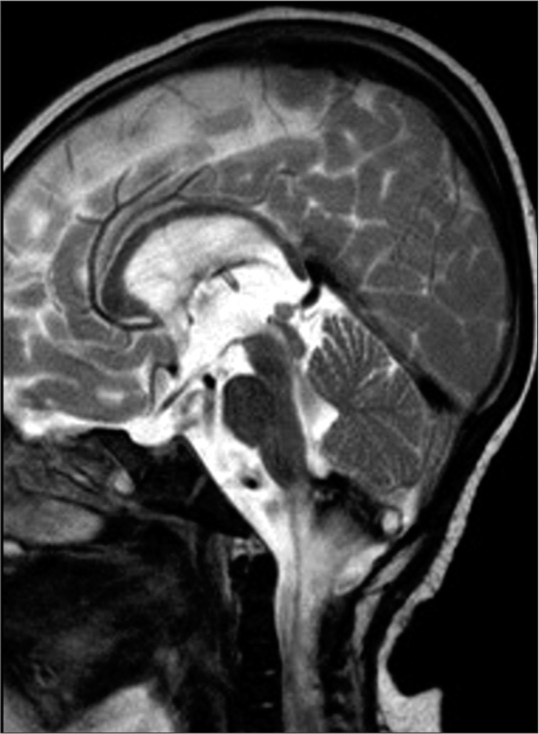
DISCUSSION
In this paper, we have presented a rare case of infantile glioma with ETV-NTRK3 fusion. This case is an ETV-NTRK3 fusion-positive tumor originating from the medullary-cervical transition zone of the medulla oblongata that grew rapidly after radiochemotherapy treatment. Cervicomedullary glioma is a rare entity, and its clinical and molecular characteristics have not been fully elucidated. To the best of our knowledge, this is the first case report of cervicomedullary glioma with ETV-NTRK3 fusion in an infant.
According to a study that performed a literature analysis of pediatric cervicomedullary gliomas,[
Due to rapid progress in gene analysis and molecular-targeted therapies for pediatric brain tumors, it has become clear that pediatric brain tumors differ from adult tumors. The frequency of ALK, ROS1, and NTRK fusion genes is relatively high in pediatric brain tumors. NTRK is an important protein for the nerve cell survival, differentiation, and death. Wu et al.[
The 2021 revision of the World Health Organization Brain Tumor Classification requires molecular genetic diagnosis to classify pediatric brain tumors. From a histopathological point of view, our case shows malignant findings with a mitotic figure and increased proliferative capacity in the small specimen, indicating a HGG. Based on NTRK gene alteration, the patient was diagnosed with an infant-type hemispheric glioma, NTRK-altered, despite being located in the brainstem. Infant-type hemispheric glioma is characterized by high-grade cellular astrocytoma arising in early childhood, mostly in the 1st year of life. The tumor is commonly located in the supratentorial region with a large mass and occasionally involves the adjacent leptomeninges. Despite the fact that this entity is named “hemispheric” glioma, fusions in the NTRK gene are not uncommon in brainstem glioma among infants. Although the characteristics and prognosis of NTRK-altered infant glioma have not been fully clarified, it seems more appropriate to categorize this tumor as an infant-type glioma, rather than an infant-type “hemispheric” glioma.
In our case, symptoms progressed and the tumor grew immediately after the induction of chemoradiotherapy. It is difficult to distinguish between tumor progression and an acute response to the treatment of the tumor. The rapid deterioration of the symptoms during chemoradiotherapy could be caused by tumor progression or an acute reaction to the treatment. Because the deterioration occurred immediately after the induction of chemotherapy and radiotherapy in this case, it is more likely a side effect of either chemotherapy, radiotherapy, or both. The exact mechanism underlying the early and delayed changes after radiation therapy in brain tumors remains unclear. According to a pathology report,[
Radiotherapy for infants should be avoided in concerns with late sequelae including cognitive dysfunction. Initially, we planned a chemotherapy-based strategy and delayed the need for radiotherapy. However, symptoms rapidly progressed after the induction of chemotherapy and the tumor was unresectable. Asheley et al.[
The increasing number of cases has resulted in the widespread use of molecular diagnosis and treatment with molecular-targeted drugs with favorable results.[
CONCLUSION
We reported a rare case of infantile glioma at the cervicomedullary region with ETV-NTRK3 fusion- and H3K27M wildtype-positivity. Recent advancement in genetic analysis enabled us to establish a new classification of brain tumors based on molecular profile. However, the characteristics of pediatric gliomas, including each molecular subtype, are still unknown, and further, research is required to clarify the prognosis and sensitivity against treatment of each tumor type. These details of our case will be useful for tumor reclassification and establishing precision treatment for this rare type of tumors.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
This work was supported by JSPS KAKENHI Grant Number 21K09093.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author(s) confirms that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing or editing of the manuscript and no images were manipulated using the AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Andrews JP, Coleman C, Hastings C, Sun PP. Oncogenic NTRK fusion in congenital spinal cord glioblastoma: Sequencing directs treatment. Lancet. 2021. 398: 2185
2. Ashley DM, Merchant TE, Strother D, Zhou T, Duffner P, Burger PC. Induction chemotherapy and conformal radiation therapy for very young children with nonmetastatic medulloblastoma: Children’s Oncology Group study P9934. J Clin Oncol. 2012. 30: 3181-6
3. Borgmann K, Haeberle D, Doerk T, Busjahn A, Stephan G, Dikomey E. Genetic determination of chromosomal radiosensitivities in G0-and G2-phase human lymphocytes. Radiother Oncol. 2007. 83: 196-202
4. Chim ST, Sanfilippo P, O’Brien TJ, Drummond KJ, Monif M. Pretreatment neutrophil-to-lymphocyte/monocyte-to-lymphocyte ratio as prognostic biomarkers in glioma patients. J Neuroimmunol. 2021. 361: 577754
5. Dahl NA, Donson AM, Sanford B, Wang D, Walker FM, Gilani A. NTRK fusions can co-occur with H3K27M mutations and may define druggable subclones within diffuse midline gliomas. J Neuropathol Exp Neurol. 2021. 80: 345-53
6. Doz F, van Tilburg CM, Geoerger B, Højgaard M, Øra I, Boni V. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol. 2022. 24: 997-1007
7. König D, Hench J, Frank S, Dima L, Bratic Hench I, Läubli H. Larotrectinib response in NTRK3 fusion-driven diffuse high-grade glioma. Pharmacology. 2022. 107: 433-8
8. . Report of brain tumor registry of Japan (1984-2000). Neurol Med Chir (Tokyo). 2009. 49: Ps1-96
9. Torre M, Vasudevaraja V, Serrano J, DeLorenz M, Malinowski S, Blandin AF. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun. 2020. 8: 107
10. Trezza A, de Laurentis C, Biassoni V, Carrabba GG, Schiavello E, Canonico F. Cervicomedullary gliomas in pediatric age: A systematic review of the literature and tertiary care center experience. Pediatr Neurosurg. 2022. 57: 149-60
11. Turesson I, Nyman J, Holmberg E, Odén A. Prognostic factors for acute and late skin reactions in radiotherapy patients. Int J Radiat Oncol Biol Phys. 1996. 36: 1065-75
12. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014. 46: 444-50

