

- Theoretical Neuroscience Research, Ridgeland, Mississippi,
- Clinical Professor of Surgery (Neurosurgery, ret.) and Adjunct Professor of Medical History (ret.), Mercer University School of Medicine, United States.
Correspondence Address:
Russell Blaylock, Theoretical Neuroscience Research, Ridgeland, Mississippi, United States.
DOI:10.25259/SNI_1007_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Russell L. Blaylock1, Miguel Faria2. New concepts in the development of schizophrenia, autism spectrum disorders, and degenerative brain diseases based on chronic inflammation: A working hypothesis from continued advances in neuroscience research. 08-Nov-2021;12:556
How to cite this URL: Russell L. Blaylock1, Miguel Faria2. New concepts in the development of schizophrenia, autism spectrum disorders, and degenerative brain diseases based on chronic inflammation: A working hypothesis from continued advances in neuroscience research. 08-Nov-2021;12:556. Available from: https://surgicalneurologyint.com/surgicalint-articles/11218/
Date of Submission
04-Oct-2021
Date of Acceptance
05-Oct-2021
Date of Web Publication
08-Nov-2021
Abstract
This paper was written prompted by a poignant film about adolescent girl with schizophrenia who babysits for a younger girl in an isolated cabin. Schizophrenia is an illness that both authors are fascinated with and that they continue to study and investigate. There is now compelling evidence that schizophrenia is a very complex syndrome that involves numerous neural pathways in the brain, far more than just dopaminergic and serotonergic systems. One of the more popular theories in recent literature is that it represents a hypo glutaminergic deficiency of certain pathways, including thalamic ones. After much review of research and study in this area, we have concluded that most such theories contain a number of shortcomings. Most are based on clinical responses to certain drugs, particularly antipsychotic drugs affecting the dopaminergic neurotransmitters; thus, assuming dopamine release was the central cause of the psychotic symptoms of schizophrenia. The theory was limited in that dopamine excess could only explain the positive symptoms of the disorder. Antipsychotic medications have minimal effectiveness for the negative and cognitive symptoms associated with schizophrenia. It has been estimated that 20–30% of patients show either a partial or no response to antipsychotic medications. In addition, the dopamine hypothesis does not explain the neuroanatomic findings in schizophrenia.
Keywords: Glutamate receptors, Immunoexcitotoxicity, Microglia, Pro-inflammatory cytokines
HISTORICAL BACKGROUND AND CLINICAL PRESENTATION
The following is a short introduction to the historical background of this disease and its clinical symptomatology. Schizophrenia was originally named dementia praecox for “premature dementia.” In 1893, the German psychiatrist Emil Kraepelin separated the two psychoses with which this disorder had been confused: dementia praecox and manic depression. It was renamed schizophrenia by the Swiss psychiatrist Eugen Bleuler in 1908, meaning in Latin “split mind or split personality.”[
As one of us has written elsewhere, the two of the leading lights of psychiatry at the turn of the century, Austrian psychoanalyst Sigmund Freud (1856–1939) and German psychiatrist Emil Kraepelin (1856–1926) had conflicting approaches to mental illness, including schizophrenia. Freud recommended psychotherapy, which was almost always unsuccessful and unfeasible in severely mentally ill patients. Kraepelin, in contrast, preferred more aggressive intervention with electroconvulsive therapy and insulin shock therapy, the former often ineffective, the latter dangerous and no longer used. Thus, psychosurgery came into vogue; fortunately, it was soon supplanted by psychotherapy, which has proven much safer and most efficacious effective in Schizophrenia in at least 75% of patients.[
Necessarily in the original descriptions of the disease, the behavioral and sociological aspects of schizophrenia have been emphasized, but not it’s anatomic, biochemical, or pathophysiological substrates. For example, schizophrenia has been described as a functional mental illness that begins gradually, occasionally almost suddenly, striking in adolescence or young adulthood. No standard neuroimaging techniques disclosed any definitive pathological abnormalities in these patients. Moreover, there was no objective pathognomonic test that would confirm the illness, which is still diagnosed on the clinical symptomatology and observed behavior.
In his autobiography, Nobel-prize winner Eric R Kandel explains how, as a psychiatrist and research neuroscientist, he had attempted to apply the scientific method to psychiatry as a new “science of the mind.” He asserts that the human mind can be studied with biological tools to create this new science. He further asserts that in time all mental disorders, including those categorized as “functional” (or psychological, including schizophrenia by implication), will be found to have a structural, biochemical, and/or molecular basis, and that the old subjective criteria for psychiatric illnesses will completely give way to the new biological and scientific “science of the mind.”[
In medical school and in our psychiatry rotations, we learn about the four fundamental symptomatologies of schizophrenia, originally described by Dr. Bleuler as the four as of schizophrenia: (1) looseness of associations, or disordered associations with a loss of contact with reality; (2) autism, a disordered conception of the world with a preference for fantasy rather than reality; (3) a disorder of affect, or an abnormal emotional state or mood; (4) ambivalence, a mixed feeling about a subject matter — one may be unconscious, but the contradictory attitudes may be indirectly expressed. Schizophrenia is also characterized by cognitive impairments, delusions, and hallucinations that are most frequently auditory.[
Neurophysiological disorders of the central nervous system neurotransmission and biochemical defects of neurotransmitters production, transport, reuptake, blockage, and degradation have provided the best theories for explaining the good to excellent responses in schizophrenic patients to a variety of neuropharmacological agents. In addition, defects in working memory associated with disconnection of the hippocampal formation with the prefrontal cortex and in neurotransmission in the dorsolateral prefrontal cortex and frontotemporal disconnection have implicated both the frontal and temporal lobes in the neuropathology of schizophrenia.[
IMMUNOEXCITOTOXICITY IN NEURODEGENERATIVE DISEASES AND SCHIZOPHRENIA
The leading author has written a number of papers on a newer hypothesis of neurodegenerative disease, of which schizophrenia is one.[
Inflammation in the brain, especially if prolonged, triggers excitotoxicity, which over time destroys, first synaptic connections, axons, and then eventually neuronal cell bodies — something seen in postmortem examinations of autistic brains.[
Both postmortem and most in vivo positron emission tomography (PET) scanning have demonstrated microglial activation in the affected areas of the brain of schizophrenia patients and autism spectrum disorder (ASD) patients.[
Interestingly, the areas of the brain most affected in schizophrenia have been shown to have higher densities of microglia than are normally seen in healthy brains (28% higher in frontal cortex and 57% higher in temporal area).[
Microglial activation and resulting immunoexcitotoxicity, especially with priming, would explain the dysfunctional behavioral control and cognitive deficiency seen in schizophrenia as this process would alter dendritic and synaptic pruning, and also interfere with later plasticity. The studies of first episode schizophrenia patients have shown extensive network damage in drug treatment naïve patients, which is also seen despite antipsychotic drug treatment.[
Microglia priming is also a critical process in this disorder and most neurodegenerative diseases.[
Activation of microglia in schizophrenia was first reported in 1999, where microglial activation was seen in the frontal cortex and hippocampus in 14 patients.[
One of the main problems we find with this theory is that there is compelling evidence that immune/inflammatory events occurring in utero and during early postnatal development can increase the risk of schizophrenia later in life, usually around adolescence.[
ASDS AND SCHIZOPHRENIA: A POSSIBLE LINK
It is also important to appreciate that schizophrenia and ASDs share core symptoms and overlap in many ways pathologically, mainly by extensive microglial activation, anatomical changes, and similar behavioral attributes.[
In addition, there appears to be a strong genetic link associated with both, and interestingly, a number of these genes have to do with control of microglial function (TREM2, TLRs, TYRO proteins, etc.).[
Research has clearly shown that early life events can have lasting impacts in the brain and behavioral function throughout life.[
Of the pro-inflammatory cytokines involved, IL-6 appears to play the major role. The studies have shown that blocking IL-6, using genetic or pharmacologic methods, prevented the long-term anatomical, and behavioral consequences of exposure to Poly I: C.[
It has also been shown that animals born to mothers who sustained an immune challenge during gestation demonstrated a specific set of abnormalities in brain function, such as deficits in working memory, abnormal executive function, impaired discrimination, and deficits in both spatial and non-spatial information processing.[
While there are many similarities between autism and schizophrenia, there are also major differences, such as excessive brain growth in the early stages of ASDs. This tends to disappear over time as the disorder progresses. One major difference is that with ASDs is that with ASDs brain inflammation is long-term and continuous, extending into adulthood.[
INFLAMMATION AND SCHIZOPHRENIA
The studies have shown that patients with recent onset schizophrenia demonstrate activation of pro-inflammatory networks and inflammatory mediators.[
It has also been shown that success in treatment parallels lowering of these inflammatory cytokines.[
We have seen that experimental studies support the link between inflammation, elevated cytokines (especially IL-6) and the development of schizophrenia, including the anatomical and pathological changes seen in the disorder. The source of the pro-inflammatory cytokines appears to be mainly from activated and or primed microglia and invading macrophages, which once in the brain take on the appearance and function of microglia.[
THE GLUTAMATE HYPOTHESIS FOR SCHIZOPHRENIA
Stone et al. noted that the dopaminergic hypothesis did not adequately explain all the neuroanatomical and clinical findings in schizophrenia patients.[
Initially, it was assumed that schizophrenia was a disorder of deficient glutamate receptor function universally. Subsequent studies came to a different conclusion. Most important, it was observed that both phencyclidine and ketamine were selective NMDA receptor blockers. Further studies also demonstrated that rather than low levels of glutamate in the brain, one sees elevated levels, particularly in the striatum and prefrontal cortex (especially anterior cingulate) following NMDA receptor blockade.[
Additional evidence comes from treatment studies which have shown that unmedicated schizophrenia patients have elevated brain glutamate levels and that once successfully treated the glutamate levels return to normal. Clinical improvement parallels the fall in striatal glutamate levels.[
That is, higher glutamate levels were seen in treatment resistant patients than in those who responded well to treatment.
The activated microglia are the main source of elevated glutamate. Inflammatory activation of microglia is accompanied by the release of excitotoxic levels of glutamate and other excitotoxic molecules such as quinolinic acid and aspartate.[
HOW NMDA RECEPTOR UNDERACTIVATION RESULTS IN IMMUNOEXCITOTOXICITY AND ELEVATED BRAIN GLUTAMATE LEVELS
Initially, when impaired NMDA receptor function was discovered in schizophrenia, it was assumed that reduced overall glutaminergic function was responsible for the negative symptoms. Recent studies have demonstrated another mechanism. Rather than a general reduction in NMDA receptor function, new studies suggest the thalamus is the main site of NMDA receptor hypofunction.[
The extraneuronal surge of glutamate occurring with NMDA receptor antagonism causes neurodegeneration most likely by acting through AMPA/kainate receptors, in particular, the calcium sensitive GluR2-lacking AMPA receptors.[
IMMUNOEXCITOTOXIC NEURODEGENERATION IN SCHIZOPHRENIA
A combination of microglial activation and suppression of GABAnergic activity by NMDA receptor suppression, leads to a significant elevation in extraneuronal glutamate levels[
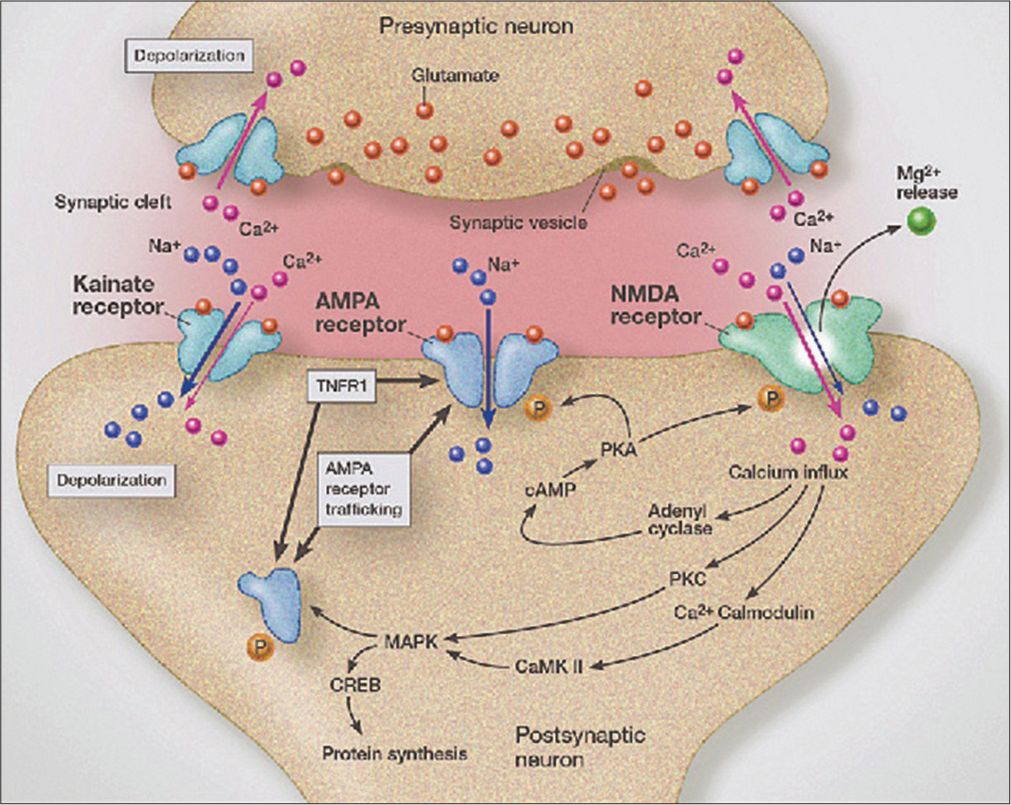
It has been hypothesized that the excitotoxicity occurs through excessive activation of AMPA/kainate receptors by the elevated extraneuronal glutamate[
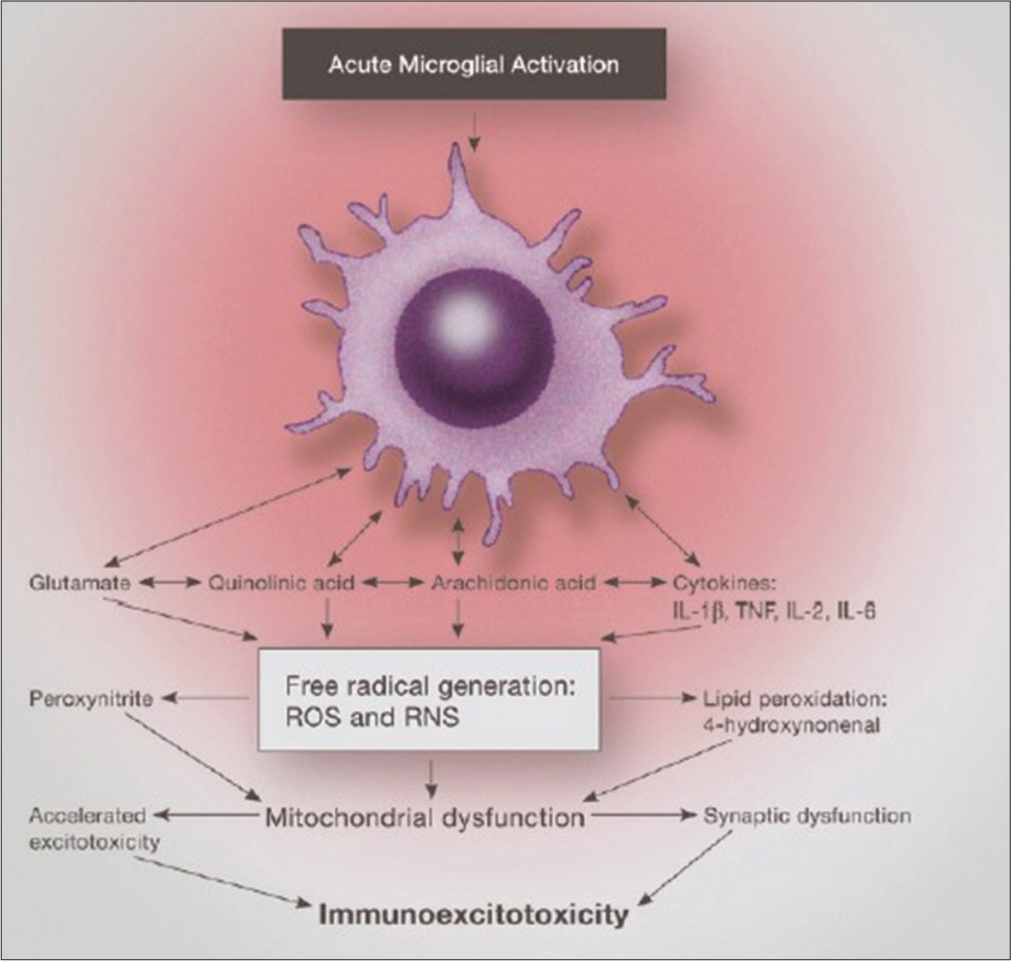
Figure 2:
Illustration of the mechanism of immunoexcitotoxicity focusing on a combination of immune activation and excitotoxicity originating from activated microglia. Once fully activated, the microglia releases a combination of immune mediators and excitotoxic compounds with subsequent generation of high levels of reactive oxygen and nitrogen species and lipid peroxidation products. This combination results in mitochondrial dysfunction, further energy deficits, and accelerated excitotoxicity with subsequent neurodegeneration of surrounding structures.
INTERACTION BETWEEN THE IMMUNE SYSTEM AND GLUTAMATE RECEPTORS: MECHANISM OF IMMUNOEXCITOTOXICITY
The earliest reports demonstrating an enhancement of excitotoxicity by TNF-alpha were by Gelbard et al. in which they used human neuronal cultures exposed to subtoxic dose of TNF-alpha and AMPA.[
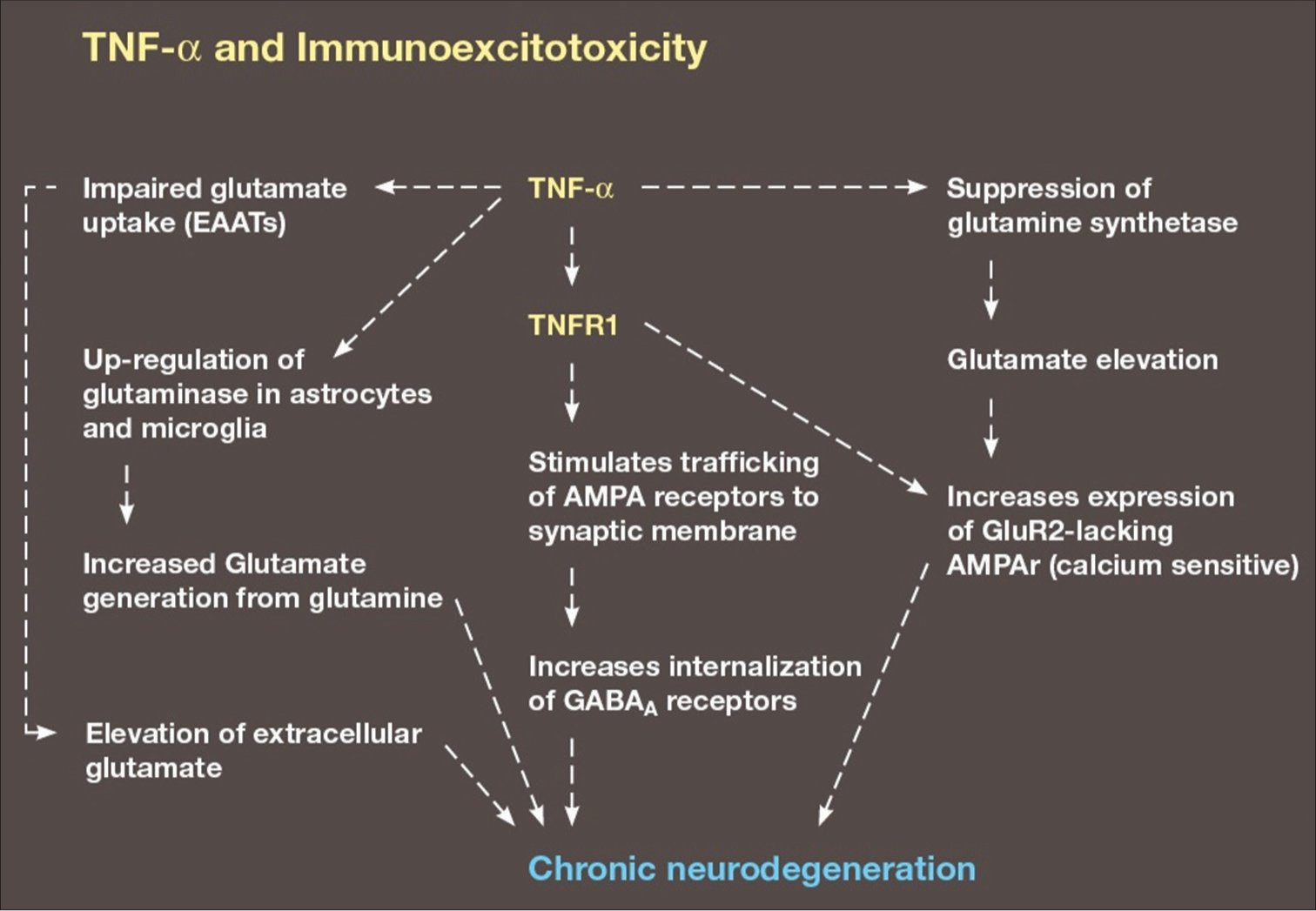
Figure 3:
Illustration demonstrating the various ways TNF-alpha enhances excitotoxicity. Other pro-inflammatory cytokines also enhance excitotoxicity, such as IL-1, IL-6, and IL-17, but TNF-alpha is the most prominent player. Excitotoxicity appears to be the final and most destructive event triggered by inflammation and/or microglial priming/activation.
It has also been shown that elevation of proinflammatory cytokines, especially TNF-alpha, inhibits the glutamate reuptake transporters GLAST and GLT-1, which raises extracellular glutamate levels to neurotoxic levels and prevents lowering of extracellular glutamate during activity of the cystine-glutamate antiporter.[
Together, these TNF-alpha related effects on glutamate receptors, enzymes and trafficking enhance excitotoxicity and intimately link inflammatory mediators to excitotoxicity. Because activated microglia are the principal source of both excitotoxins and inflammatory mediators, it becomes difficult to determine exactly how much each contributes to neurodegeneration. My impression is that excitotoxicity is the final common pathway responsible for most of the neurodestruction when microglial are activated.[
It appears that the neurological damage occurs first either during the third trimester of pregnancy or soon after birth and that until the symptoms of psychosis present themselves. There is a progressive interference with neurodevelopment as well as a process of progressive neurodegeneration of the most involved areas of the brain following birth.[
NEURODEGENERATION AND SCHIZOPHRENIA
Another important suggestion of excitotoxic neurodegeneration occurs with the widespread loss of neurons and connectivity as the disorder progresses. One sees progressive loss of grey matter volume beginning early in life which continues chronically.[
Of interest, blocking agonist of metabolic glutamate receptor types 2 and 3 blocks the neurotoxic effects of NMDA antagonists in preclinical models of schizophrenia.[
It has been suggested that schizophrenics are generally heavy smokers.[
SCHIZOPHRENIA AND GUT INFLAMMATION
The big question is: What is causing the chronic, low-level inflammation? It has been shown clearly that prenatal infections in the mother can cause postnatal schizophrenia and more recent studies using non-infectious Poly I: C have shown that the mechanism involves cytokine elevations, principally IL-6 and not actual infection.[
Another link that has a lot of validity, certainly in some cases of both ASD and schizophrenia, is gut inflammation. Gliadin and gluten have been shown to trigger chronic microglial activation and are linked clinically to a number of cases of schizophrenia.[
As for why some cases fail to improve, it has been shown that gluten can trigger increased gut permeability, which can persist in some cases after starting a gluten-free diet, as there are often other contributing factors also linked to gut permeability, such as use of nonsteroidal anti-inflammatory drugs. Translocation of other food proteins and colon/intestinal bacteria can trigger continued microglial activation with resulting persistent immunoexcitotoxicity. In addition, gut inflammation can send afferents through the vagus nerve that activate brain microglia.[
CONCLUSION
It is of interest that many of the antipsychotic medications used to treat schizophrenia are known to suppress microglial activation and alter glutamate receptor function.[
As for the neurotransmitters, especially dopamine, it has been shown that treatment resistant forms of schizophrenia (type B patients) were associated with relatively normal levels of dopamine synthesis in the striatum and elevated glutamate levels in the anterior cingulate cortex.[
The disruption of several neurotransmitters in schizophrenia is consistent with immunoexcitotoxicity, as a number of neuron types, receptor types and subtypes are affected by high levels of inflammation and excitotoxicity, with associated elevations in reactive oxygen and nitrogen species and lipid peroxidation — that is, these changes are epiphenomenon.
In our opinion, we should be addressing the central mechanism of the problem (immunoexcitotoxicity and microglial activation) rather than attempting to fine tune neurotransmitter disruptions, which can appear in a complex, variable, and often confusing presentation. This also requires attention to gut inflammation and correction of the microbiome.[
*An AMPA receptor is the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor is an ionotropic transmembrane receptor for glutamate that mediates fast synaptic transmission in the central nervous system (CNS), and it is considered a non-NMDA receptor
Dr. Russell L. Blaylock is an Associate Editor-in-Chief of the Neuroinflammation and Neuropsychiatry sections and a Consulting Editor in Basic Neuroscience for Surgical Neurology International (SNI).
Dr. Miguel A. Faria is an Associate Editor in Chief in Neuropsychiatry; History of Medicine; and Socioeconomics, Politics, and World Affairs of Surgical Neurology International (SNI).
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abbott C, Bustillo J. What have we learned from proton magnetic resonance spectroscopy about schizophrenia? A critical update. Clin Neuropharmacol. 2001. 24: 43-9
2. Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D. Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry. 1999. 156: 1646-9
3. Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: Support for hyperglutamatergic effect of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000. 57: 270-6
4. Arrango C, Rapado-Castro M, Reig S, Castro-Fornieles J, Gonzales-Pinto A, Ptero S, Baeza I. Progressive brain change in children and adolescent with first-episode psychosis. Arch Gen Psychiatry. 2012. 69: 16-26
5. Arrode-Bruses G, Bruses JL. Maternal immune activation by poly (I: C) induces expression of cytokines IL-1ß and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J Neuroinflammation. 2012. 9: 83
6. Ashdown H, Dumont Y, Ng M, Poole S, Boska P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection in the fetus: Implications for schizophrenics. Mol Psychiatry. 2006. 11: 47-55
7. Auer RN. Effect of age and sex on N-methyl-D-aspartate antagonists-induced neuronal necrosis in rats. Stroke. 1996. 27: 743-6
8. Bale TL, Baram TZ, Brown AS, Goldsstein JM, Insel TR, McCarthy MM. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010. 68: 314-9
9. Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. 1999. 271: 126-8
10. Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Rev. 1998. 9: 259-75
11. Bian Q, Kato T, Monji A, Hashioka S, Mizoquchi Y, Horikawa H. The effect of atypical antipsychotics, perospirone, zoprasiclone and quetrapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008. 32: 42-8
12. Bilbo SD, Schwartz JM. Early-life programming of later-life brain and behavior: A critical role for the immune system. Front Behav Neurosci. 2009. 3: 14
13. Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-a unifying hypothesis. Surg Neurol Int. 2011. 2: 107
14. Blaylock RL. Immunology primer for neurosurgeons and neurologists Part 2: Innate brain immunity. Surg Neurol Int. 2013. 4: 118
15. Blaylock RL. Parkinson’s disease: Microglial/macrophage-induced immunoexcitotoxicity as a central mechanism of neurodegeneration. Surg Neurol Int. 2017. 8: 65
16. Blaylock RL, Strunecka A.editors. The Cerebellum in autism spectrum disorders. Cellular and Molecular Biology of Autism Spectrum Disorders. United Arab Emirates: Bentham Books; 2010. p. 17-31
17. Bora E, Fornito A, Radua J, Walterfang M, Seal M, Wood SJ. Neuroanatomical abnormalities in schizophrenia: A multimodal voxelwise meta-analysis and meta-regression analysis. Schizophr Res. 2011. 127: 46-57
18. Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensomotor gating in adult rats, Implications for the etiopathogenesis of schizophrenia. Neuropsychpharmacology. 2002. 26: 204-15
19. Bridges RJ, Natale NR, Patel SA. System xc-cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012. 165: 20-34
20. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: A review of epidemiological and translational studies. Am J Psychiatry. 2010. 167: 261-80
21. Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004. 161: 889-95
22. Burns TM, Clough JA, Klein RM, Wood GW, Bergman NE. Developmental regulation of cytokine expression in the mouse brain. Growth Factors. 1993. 9: 253-8
23. Bustos G, Abarca J, Forray MI, Gysling K, Bradberry CW, Roth RH. Regulation of excitatory amino acid release by N-methyl-D-aspartate receptors in rat striatum: In vivo micro-dialysis studies. Brain Res. 1992. 585: 105-15
24. Carter K, Dickerson J, Schoepp DD, Reilly M, Herring N, Williams J. The mGlu2/3 receptor agonists LY379268 injected into cortex or thalamus decreases neuronal injury in retrosplenal cortex produced by NMDA receptor antagonist MK-801: Possible implication for psychosis. Neuropharmacology. 2004. 47: 1135-45
25. Cheng C, Yu K, Fung G, Leung M, Wong C, Li Q. Autistic disorder and schizophrenia: Related ort remote? An anatomical likelihood estimation. PLoS One. 2010. 5: e12233
26. Chenz Z, Deng W, Gong Q, Huang C, Jiang L, Li M. Extensive brain structural network anomality in first-episode treatment-naïve patients with schizophrenia: Morphometrical and covariation study. Psychol Med. 2014. 44: 2489-501
27. Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007. 36: 361-5
28. Colibazzi T, Wexler BE, Bansal R, Hao X, Liu J, Sanchez-Pena J. Anatomical abnormalities in gray and white matter of the cortical surface in persons with schizophrenia. PLoS One. 2013. 8: e55783
29. Connor CM, Crawford BC, Akbarian S. White matter neuron alterations in schizophrenia and related disorders. In J Dev Neurosci. 2011. 29: 325-34
30. Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci. 2003. 1003: 318-27
31. da Cunha A, Nunes FB, Lunardelli A, Pauli V, Amaral RH, de Oliveira LM. Treatment with N-methyl-D-aspartate receptor antagonist (MK-801) protects against oxidative stress in lipopolysaccharide-induced acute lung injury. Int Immunopharmacol. 2011. 11: 706-11
32. Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997. 42: 1-8
33. de la Fuente-Sandoval C, Leon-Ortez P, Azacarraga M, Stephano S, Favila R, Diaz-Galvis L. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first episode psychosis: A longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry. 2013. 70: 1057-66
34. de la Funte-Sandoval C, Leon-Ortez P, Favila R, Stephanos S, Mamo D, Ramirez-Bermudez J. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first episode psychosis. Neuropsychopharmacology. 2011. 36: 1781-91
35. Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dynamic function. Biol Psychiatry. 2014. 75: e11-3
36. Dennison U, McKernan D, Cryan J, Dianan T. Schizophrenia patients with a history of childhood trauma have a pro-inflammatory phenotype. Psychol Med. 2012. 42: 1865-71
37. Deutsh SI, Rosse RB, Schwartz BL, Mastropaolo JA. A revised excitotoxic hypothesis of schizophrenia: Therapeutic implications. Clin Neuropharmacol. 2001. 24: 43-9
38. Ergun C, Urhan M, Ayer A. A review on the relationship between gluten and schizophrenia: Is gluten the cause?. Nutr Neurosci. 2018. 21: 455-66
39. Farber NB, Wozniak DF, Price MT, Labruyere J, Huss J, St Peter H. Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: Potential relevance to schizophrenia?. Biol Psychiatry. 1995. 38: 788-96
40. Farber NB. The NMDA receptor hypofunction model of psychosis. Ann N Y Acad Sci. 2003. 1003: 119-30
41. Faria MA. Schizophrenia in the Dramatic Film “Occupied” (2011) and its Critics. Available from: https://www.haciendapublishing.com/schizophrenia-in-the-dramatic-film-occupied-2011-and-its-critics-by-miguel-a-faria-md. [Last assecced on 2021 Oct 04].
42. Faria MA. Violence, mental illness, and the brain-a brief history of psychosurgery: P art 1-from trephination to lobotomy. Surg Neurol Int. 2013. 4: 49
43. Floresco SB, Todd CL, Grace AA. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001. 21: 4915-22
44. Galtieri DJ, Estep CM, Wokosin DL, Traynelis S, Surmeier DJ. Peduculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife. 2017. 6: e30352
45. Gelbard HA, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: Implications for AIIDS neuropathogenesis. Dev Neurosci. 1994. 16: 417-22
46. George M, Amruthesehwar R, Rajkumar RP, Kattimani D, Dkhar SA. Newer antipsychotics and upcoming molecules for schizophrenia. Eur J Clin Pharmacol. 2013. 69: 1497-509
47. Gober R, Ardalan M, Shiadeh SM, Duque L, Garamsregi SP, Ascona M. Microglial activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Path. 2021. 1: e13003
48. Godbout JP, Moreau M, Lestage J, Sparkman NL, O’Connor J, Castanon N. Aging exacerbates depressive-like behavior in mice in response to activation if the peripheral innate immune system. Neuropsychopharmacology. 2007. 33: 2341-51
49. Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor alpha induces cFOS and strongly promotes glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001. 8: 590-9
50. Holmes S, Hinz R, Drake R, Gregory C, Conen S, Matthews J. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: A [11C](R)-PK11195 positron emission tomography study. Mol Psychiatry. 2016. 21: 1672-9
51. Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: An update for the 21st Century. J Psychopharmacology. 2015. 29: 97-115
52. Howes OD, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl Psychiatry. 2017. 7: e1024
53. Hutshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull. 2008. 34: 354-66
54. Ikonomidou C, Bosch F, Miksa M, Bittgau P, Vockler J, Dirkranian K. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999. 283: 70-4
55. Iltis I, Koski DM, Eberly LE, Lenson CD, Deelehand DK, Valette J. Neurochemical changes in the rat prefrontal cortex following acute phencyclidine treatment: An in vivo localized (1)H MRS study. NMR Biomed. 2009. 22: 737-44
56. Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D. Has an angel shown the way? Etiology and therapeutic implications of the PCP/NMDA model schizophrenia. Schizophr Bull. 2012. 38: 958-66
57. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991. 148: 1301-8
58. Kandel ER. Search of Memory: The Emergence of a New Science of Mind. Available from: https://www.surgicalneurologyint.com/wp-content/uploads/2020/08/10205/sni-11-252.pdf. [Last assecced on 2021 Oct 04].
59. Kato T, Monji A, Hashioka S, Kanba S. Risperidone significantly inhibits interferon-gamma induced microglial activation in vitro. Schizophr Res. 2007. 92: 108-15
60. Kegelos LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2012. 69: 449-59
61. Kigerl KA, Ankeny DP, Garg SK, Guan Z, Lai W, McTigue DM. System X (c) (-) regulates microglia and macrophage glutamate excitotoxicity in vivo. Exp Neurol. 2012. 233: 333-41
62. Kimura M, Toth LA, Agostini H, Cady AB, Majde JA, Kruger JM. Comparison of acute phase responses induced in rabbits by lipopolysaccharide and double-stranded RNA. Am J Physiol. 1994. 267: R1596-605
63. Kiss T, Hoffmann WE, Scott L, Kawabe TT, Milici AJ, Nilsen EA. Role of thalamic projections in NMDA receptor-induced disruption of cortical slow oscillation and short-term plasticity. Front Psychiatry. 2011. 2: 14
64. Kolvin I. Studies in the childhood psychoses I. Diagnostic criteria and classification. Br J Psychiatry. 1971. 118: 381-4
65. Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995. 13: 9-19
66. Laskaris LE, Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E. Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol. 2016. 173: 666-80
67. Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008. 28: 2119-30
68. Levinta A, Mukovozov I, Tsoutsoulas C. Use of a gluten-free diet in schizophrenia: A systematic review. Adv Nutr. 2018. 9: 824-32
69. Liao SL, Chen CJ. Differential effects of cytokines and redox potential on glutamate uptake in rat cortical glial cultures. Neurosci Lett. 2001. 299: 113-6
70. Libby JE, Sweeten TL, McMahon WM, Funjinami RS. Autistic disorder and viral infections. J Neurovirol. 2005. 11: 1-10
71. Lohr JB, Flynn K. Smoking and schizophrenia. Schizophr Res. 1992. 8: 93-102
72. Meyer U, Feldon J, Yee BK. A review of the fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009. 35: 959-72
73. Meyer UR, Feldon J, Dammann O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation?. Pediatr Res. 2011. 69: 26R-33R
74. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alteration in schizophrenia: Clinical status and antipsychotic effects. Biol Psychiatry. 2011. 70: 663-71
75. Moghaddam B, Adam B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruption associated with the prefrontal cortex. J Neurosci. 1997. 17: 2921-7
76. Monji A, Kato T, Kanba S. Cytokines and schizophrenia: Microglial hypothesis of schizophrenia. Psychiatry Clin Neurosci. 2009. 63: 257-65
77. Muller N. Inflammation in schizophrenia: Pathologenetic aspects and therapeutic considerations. Schizophr Bull. 2018. 44: 973-82
78. Muller N, Schwartz MJ. The immunological basis of glutamatergic disturbance in schizophrenia: Towards an integrated view. J Neural Transm Suppl. 2007. 72: 269-80
79. Nak S, Kim YK. Monocytic, Th1 and Th2 cytokine alterations in the pathophysiology of schizophrenia. Neuropsychobiology. 2007. 56: 55-63
80. Nyffer M, Meyer U, Yee BK, Feldon J, Knuesel I. Maternal immune activator during pregnancy increases limbic GABAA receptor immunoreactivity in the adult offspring: Implications in schizophrenia. Neuroscience. 2006. 143: 51-62
81. Nyuygen D, Alvi MV, Kim KY, Kang T, Scott RT, Noh YH. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011. 1: e240
82. Okada K, Hashimoto K, Iwata Y, Nakamura K, Tsujii M, Tsuchiya KJ. Decreased serum levels of transforming growth factor-ß1 in patients with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2007. 31: 187-90
83. Olomos G, Llado J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014. 2014: 861231
84. Parket-Athill EC, Tan J. Maternal activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neurosignals. 2010. 18: 113-28
85. Patterson PH. Immune involvement in schizophrenia and autism: Etiology, pathology, and animal models. Behav Brain Res. 2009. 205: 313-21
86. Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracy KJ. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Mol Med. 2003. 9: 125-34
87. Pinkman AE, Hopfinger JB, Pelphrey KA, Piven J, Penn DL. Neural basis for impaired social cognition in schizophrenia and autism spectrum disorders. Schizophr Res. 2008. 99: 164-75
88. Ponzio NM, Servatius R, Beck K, Marzouk A, Kreider T. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspiring. Ann N Y Acad Sci. 2007. 1107: 118-28
89. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi EInflammatory cytokine alteration in schizophrenia: A systematic quantitative review, Biol Psychiatry. 2008. 63: 801-8
90. Prata J, Santos SG, Almeida MI, Coelho R, Barbosa MA. Bridging autism spectrum disorders and schizophrenia through inflammation and biomarkers-pre-clinical and clinical investigations. J Neuroinflammation. 2017. 14: 179
91. Randel PM, McCurdy L.editors. The Psychiatry Learning System-a Multimedia Self-Instructional Course in Basic Psychiatry. Charleston, SC: Department of Psychiatry and Behavioral Sciences, Medical University of South Carolina; 1975. p.
92. Roumier A, Pascual O, Bechade C, Wakselman S, Poncer JC, Real E. Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS One. 2008. 3: e2595
93. Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: A 4-T proton MRS study. Am J Psychiatry. 2005. 162: 394-6
94. Salloway SP, Malloy PF, Cummings JF.editors. The Neuropsychiatry of Limbic and Subcortical Disorders. Washington, DC: American Psychiatric Publishing, Inc.; 1997. p. 155-66
95. Salloway SPMalloy PFDuffy JD. The Frontal Lobes and Neuropsychiatric Illness. Available from: https://www.surgicalneurologyint.com/surgicalint-articles/frontal-lobe-syndromes-in-neuropsychiatry-a-book-review. [Last assecced on 2021 Oct 04].
96. Samaroo D, Dickerson F, Kasarda DD, Green PH, Briani C, Yolken RH. Novel immune response to gluten in individuals with schizophrenia. Schizophr Res. 2010. 118: 248-55
97. Severance EG, Prandovszky E, Castiglione J. Gastroenterology issues in schizophrenia: Why the gut matters. Curr Psychiatry Rep. 2015. 17: 27
98. Sharp FR, Tomtaka M, Bernaudin M, Tomitaka S. Psychosis: Pathological activation of limbic thalamocortical circuits by psychominetics and schizophrenia?. Trends Neurosci. 2001. 24: 330-4
99. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007. 27: 10695-702
100. Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008. 15: 323-30
101. Steiner J, Walker M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission?. J Neuroinflammation. 2011. 8: 94
102. Stone JM, Morrison PD, Pilowski LS. Glutamate and dopamine dysregulation in schizophrenia-a synthesis and selective review. J Psychopharmacol. 2007. 21: 440-52
103. Strunecka A, Blaylock RL, Patocka J, Strunecky O. Immunoexcitotoxicity as the central mechanism of etiopathology and treatment of autism spectrum disorders: A possible role of fluoride and aluminum. Surg Neurol Int. 2018. 9: 74
104. Sugino H, Futamura T, Mitsumoto Y, Maeda K, Marunaka Y. Atypical antipsychotics suppress production of proinflammatory cytokines and up-regulate interleukin-10 in lipopolysaccharide-treated mice. Prog Neuropsychopharmacol Biol Psychiatry. 2009. 33: 303-7
105. Suzuki T, Remington G, Mulsant BH, Rajji TK, Uchida H, Gaff-Guerrero A. Treatment resistant schizophrenia and response to antipsychotics: A review. Schizophr Res. 2011. 133: 54-62
106. Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006. 281: 21362-8
107. Tay TL, Bechade C, D’Andrea I, St-Pierre MK, Henry MS, Roumier A. Microglia gone rogue: Impacts on psychiatric disorders across the lifespan. Front Mol Neurosci. 2018. 10: 421
108. Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007. 85: 2059-70
109. Toal F, Bloemen OJ, Deeley Q, Tunstall N, Daly EM, Page L. Psychosis and autism: Magnetic resonance imaging study if brain anatomy. Br J Psychiatry. 2009. 194: 418-25
110. Tomitaka S, Tomitaka M, Tolliver BK, Sharp FR. Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK801) injures pyramidal neurons in rat retrosplenial cirtex. Eur J Neurosci. 2000. 12: 1420-30
111. van Berckel B, Bossong MG, Boelaard R, Kloet R, Schuitmaker A, Casters E. Microglia activation in recent-onset schizophrenia: A quantitative (R)-[11C] PK11195 positron emission tomography study. Biol Psychiatry. 2008. 64: 820-2
112. van der Doef TF, de Witte LD, Sutterland AL, Jobse E, Yaqub M, Boellaard R. In vivo (R)-[(11)C] PK11195 PET imaging of 18kDa translocator protein in recent onset psychosis. NPJ Schizophr. 2016. 2: 16031
113. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005. 57: 67-81

