

- Department of Surgery, College of Medicine, University of Ibadan, Ibadan, Nigeria
- Department of Anaesthesia, College of Medicine, University of Ibadan, Ibadan, Nigeria
- Department of Haematology, College of Medicine, University of Ibadan, Ibadan, Nigeria
Correspondence Address:
Matthew T. Shokunbi
Department of Surgery, College of Medicine, University of Ibadan, Ibadan, Nigeria
DOI:10.4103/sni.sni_180_18
Copyright: © 2019 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Oluwakemi A. Badejo, Olusola K. Idowu, James A. Balogun, Wuraola A. Shokunbi, Simbo D. Amanor-Boadu, Matthew T. Shokunbi. Outcome of cranial surgery in Nigerian patients with hemoglobinopathies: A retrospective study. 06-Feb-2019;10:16
How to cite this URL: Oluwakemi A. Badejo, Olusola K. Idowu, James A. Balogun, Wuraola A. Shokunbi, Simbo D. Amanor-Boadu, Matthew T. Shokunbi. Outcome of cranial surgery in Nigerian patients with hemoglobinopathies: A retrospective study. 06-Feb-2019;10:16. Available from: http://surgicalneurologyint.com/surgicalint-articles/9200/
Date of Submission
06-Jun-2018
Date of Acceptance
18-Dec-2018
Date of Web Publication
06-Feb-2019
Abstract
Background:Surgical intervention in patients with hemoglobinopathies has been extensively reviewed in the literature, but information on the outcome of cranial surgery in this patient population in sub-Saharan Africa is limited.
Methods:This is a retrospective study of patients with hemoglobinopathies, who underwent brain surgery in our facility. The review covered a 5-year period. We examined patient- and surgery-related variables and described the surgical complications as well as the 60-day mortality.
Results:A total of nine procedures (eight under general anesthesia and one under local anesthesia) were performed on seven patients with hemoglobinopathy during the study period. Eight (88.9%) of these were done in female patients and one (11.1%) in a male patient. Six (66.7%) were performed in patients with no previous history of blood transfusion. Hb SC accounted for five (55.6%), Hb SS for three (33.3%), and Hb CC for one (11.1%) procedure, respectively. Three (33.3%) of these procedures were brain tumor-related, three (33.3%) trauma-related, one (11.1%) cosmetic, one (11.1%) vascular, and one for a postoperative complication. Only one (11.1%) procedure was associated with preoperative blood transfusion, whereas there was a need for blood transfusion following five (55.6%) of the procedures. There was a mortality rate of 11.1% (1 case). Other complications were recorded after three (33.3%) of the procedures and none with five (55.6%) of the procedures.
Conclusion:Neurosurgery is possible and safe in patients with hemoglobin disorders. Adequate preoperative preparation, proper anesthetic techniques, meticulous surgery, and excellent postoperative care can help optimize outcome of surgical intervention in this patient population.
Keywords: Blood transfusion, hemoglobinopathy, neurosurgery, sickle cell disease
INTRODUCTION
Hemoglobinopathies are autosomal recessive hematological genetic disorders characterized by production of abnormal hemoglobin. These include structural defects of the beta globin chain (hemoglobin variants) and quantitative abnormalities of alpha or beta-globin chains (thalassemia syndromes).[
This group of patients may require surgical intervention during the course of their lifetime, either as a direct consequence of their disease or from unrelated causes. The surgical management of patients with sickle cell disease poses a daunting challenge because of a variety of factors. These include low blood reserve, reduced oxygen-carrying capacity of the abnormal red blood cells under anesthesia, increased susceptibility to infections, the high tendency for thromboembolic events, and the potential for precipitating sickle cell crisis.[
METHODS
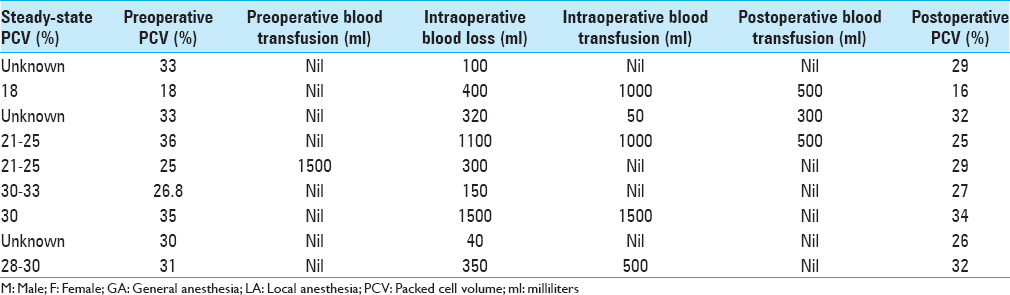
We performed a retrospective review of all patients with hemoglobinopathy, who underwent cranial neurosurgical procedures in our facility between January 1, 2013 and January 1, 2018. Data extracted from their medical records include their biodata, type of hemoglobinopathy, history of previous surgeries, comorbidities, past blood transfusion, previous sickle cell crisis, and form. Compliance with routine medications, neurosurgical diagnosis, steady-state packed cell volume, type of anesthesia, and neurosurgical procedure done were recorded. We also documented the estimated intraoperative blood loss, pre/postoperative packed cell volume, intra/postoperative blood transfusion, duration of surgery, the period of intensive care unit admission if any, length of hospitalization, complications, and outcome. These were all recorded and analyzed. The primary outcome measure was the 60-day mortality, and the secondary outcome measures were the procedure-related complications.
RESULTS
We performed 1245 neurosurgical procedures during the study period. There were seven patients with hemoglobin disorders, who had nine (0.72%) procedures during this period.
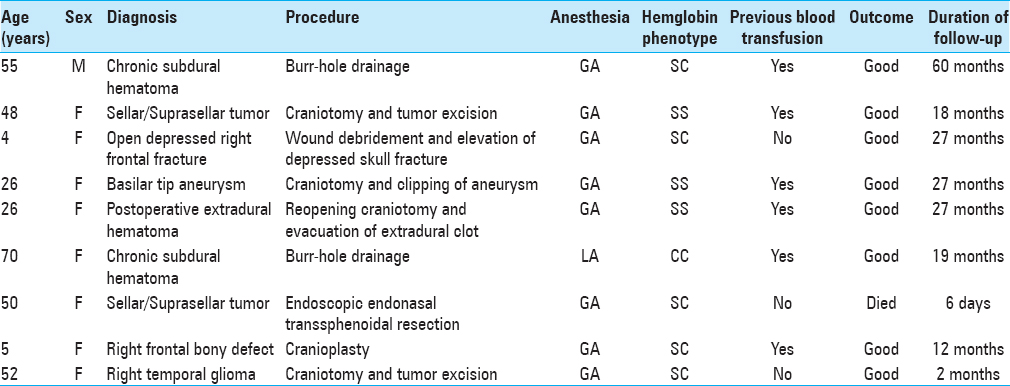
Figure 1
Right temporal tumor: axial cranial images. (a) Preoperative: cranial magnetic resonance imaging (T1, contrast-enhanced) showing lobulated, partly cystic tumor. Histologically diagnosed as anaplastic astrocytoma. (b) Postoperative: cranial computerized tomography scan (contrast-enhanced); gross near-total resection
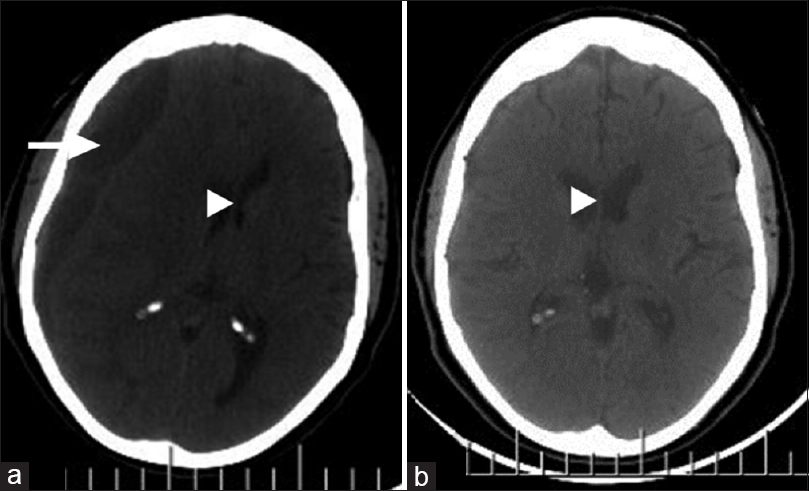
Figure 2
Right cerebral hemispheric chronic subdural hematoma; axial images, cranial computerized tomography scan. (a) Preoperative: mixed hypo- and isodense subdural collection (white arrow) with substantial mass effect (white arrow head). (b) Postoperative (6 weeks later): total evacuation of hematoma and restoration of brain midline (white arrow head)
DISCUSSION
Hemoglobinopathy remains a disease of global health importance.[
Great care must be taken to avoid factors that can predispose patients to sickle cell crisis in the preoperative period. These include hypoxia, acidosis, and hypothermia.[
Based on our experience with our patients, we suggest that preoperative workup of patients with sickle cell disease includes complete blood count, serum electrolytes, coagulation profile, and adequate hydration (to prevent vaso-occlusive crisis). Preoperative blood transfusion should be reserved for patients with hematocrits below their known steady-state levels or less than 30% in those whose steady-state levels are unknown. These patients should have preoxygenation to 100% prior to commencement of induction of anesthesia and adequate oxygenation must be maintained throughout the surgery. Intraoperative measures should include blood transfusion if blood loss exceeds 500 ml or if there is evidence of hemodynamic instability. It is important to prevent hypothermia and achieve optimal pain control in these patients to forestall sickle cell disease-related crises. Given the hypercoagulable states of these patients and the increased risk of thromboembolism, we advocate institution of nonpharmacologic deep venous thrombosis prophylaxis techniques and early ambulation when not contraindicated. Prophylactic administration of broad spectrum intravenous antibiotics and (intravenous) antibiotics coverage continued up to 48 h after surgery should be employed to prevent postoperative sepsis.
Various publications have shed light on the surgical outcomes in patients with sickle cell disease, with most of these involving orthopedic, general surgical, and otorhinolaryngological procedures.[
All the nondeath complications in our patients were successfully managed. Only one patient in our series had postoperative sepsis, which was related to a preoperative infected, plated, and malunited right femoral fracture. He subsequently had implant removal and modified Belfast procedure by the orthopedic team. The presence of comorbidities did not reflect negatively on the outcome in our series. One of the patients had vaso-occlusive crisis following debridement and elevation of an open depressed right frontal fracture, which was secondary to preoperative sepsis. She in fact had a vaso-occlusive crisis in the immediate preoperative period. The mortality was in a 50-year-old Hb SC patient, with a giant pituitary tumor who had an endoscopic transsphenoidal resection, which was terminated on account of excessive intraoperative blood loss (1500 ml). She was transfused with three units of whole blood and a unit of fresh frozen plasma. The patient's preoperative and postoperative packed cell volumes were 34% and 35%, respectively. She had transient diabetes insipidus postoperatively but remained neurologically intact. The patient had a sudden neurologic deterioration 17 h later, with no intracranial complication on cranial computerized tomography scan. This necessitated endotracheal intubation and mechanical ventilation. The neurologic status worsened progressively, and she died on the fifth-day postoperation. An autopsy revealed residual pituitary tumor, lobar pneumonia, benign nephrosclerosis, hepatomegaly, fatty liver, and splenomegaly (with acute congestion). Could the hemorrhagic complication have been caused by sickle cell hepatopathy or was it purely a surgical complication? The mortality may be as much related to the procedure as it was to the patient's sickle cell disease. The only other type of hemoglobinopathy in our study aside sickle cell disease was Hb CC, which constituted 0.08% of all cases done over the study period. At follow-up, which ranged between 2 months and 5 years, six of the patients representing eight (88.9%) of the procedures were alive and well.
CONCLUSION
Neurosurgery is possible and safe in patients with hemoglobin disorders. Proper preoperative preparation, meticulous anesthesia/surgery, and excellent postoperation care can help improve outcome.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Adewoyin AS. “Management of sickle cell disease: A review for physician education in Nigeria (Sub-Saharan Africa)”. Anemia. 2015. 2015: 791498-
2. Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci. 1998. 850: 251-69
3. Ashley-Koch A, Yang Q, Olney RS. Sickle Hemoglobin (Hb S) allele and sickle cell disease: A huge review. Am J Epidemiol. 2000. 151: 839-45
4. Babalola BO, Salman YA, Abiola AM, Okezie KO, Oladele AS. Spontaneous epidural hematoma in sickle cell anaemia: A case report and literature review. J Surg Tech Case Rep. 2012. 4: 135-37
5. Charache S, Conley CL, Waugh DF, Ugoretz RJ, Spurell JR. Pathogenesis of hemolytic anemia in homozygous hemoglobin C disease. J Clin Invest. 1961. 46: 1795-811
6. Coker NJ, Milner PF. Elective surgery in patients with sickle cell anemia. Arch Otolaryngol. 1982. 108: 574-6
7. Crotty EE, Meier ER, Wells EM, Packer RJ. Anaplastic ependymoma in a child with sickle cell anemia: A case report highlighting treatment challenges for young children with central nervous system tumours and underlying vasculopathy. Paediatr Blood Cancer. 2015. 63: 547-50
8. Diebold P, Humbert J, Djientcheu VP, Gudincht F, Rilliet B. Salmonella epidural abscess in sickle cell disease: Failure of the non-surgical treatment. J Natl Med Assoc. 2003. 95: 1095-8
9. Elisabeth K. Hemoglobinopathies: Clinical manifestations, diagnosis and treatment. Dtsch Arztebl Int. 2011. 108: 532-40
10. Estcourt LJ, Fortin PM, Trivella M, Hopewell S. Pre-operative blood transfusions for sickle cell disease. Cochrane Database Syst Rev. 2016. 4: CD003149-
11. Fisher L. Perioperative care of the patient with sickle cell disease. AORN J. 2011. 93: 150-6
12. Forget BG, Bunn HF. Classification of the disorders of hemoglobin. Cold Spring Harb Perspect Med. 2013. 3: a011684-
13. Galadanci N, Wadil BJ, Balogun TM, Ogunrinde GO, Akinsulie A, Hasan-Hanga F. Current sickle cell disease management practices in Nigeria. Int Health. 2014. 6: 23-8
14. Gross SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am J Prev Med. 2011. 41: S398-405
15. Hankinson TC, Bohman LE, Heyer G, Licursi M, Ghatan S, Feldstein MA. Surgical treatment of Moyamoya syndrome in patients with sickle cell anemia: Outcome following Encephaloduroarteriosynangiosis. J Neurosurg Pediatr. 2008. 1: 211-6
16. Koshy M, Weiner SJ, Miller ST, Sleeper LA, Vichinsky E, Brown AK. Surgery and anesthesia in sickle cell disease. Cooperative study of sickle cell diseases. Blood. 1995. 86: 3676-84
17. Kuruvath S, Basu S, Elwitigala JP, Yaneza A, Namyak SS, Aspoas AR. Salmonella enteritis brain abscess in a sickle cell disease patient: Case report and review of the literature. Int J Inf Dis. 2008. 12: 298-302
18. Kutlar F. Diagnostic approach to hemoglobinopathies. Hemoglobin. 2007. 31: 243-50
19. Lanzkron S, Carroll CP, Haywood C. Mortality rates and age at death from sickle cell disease: U.S., 1979-2005. Public Health Rep. 2013. 128: 110-6
20. Memish ZA, Saeedi MY. Six-year outcome of the national premarital screening and genetic counselling program for sickle cell disease and β-thalassemia in Saudi Arabia. Ann Saudi Med. 2011. 31: 229-35
21. Modell B, Darlison M. Global epidemiology of hemoglobin disorders and derived service indicators. Bull World Health Organ. 2008. 86: 480-7
22. Oyesiku NM, Barrow DL, Eckman JR, Tindall SC, Colohan AR. Intracranial aneurysms in sickle-cell anemia: Clinical features and pathogenesis. J Neurosurg. 1991. 75: 356-63
23. Patrinos G, Giardine B, Riemer C, Miller W, Chui DHK, Anagnou NP. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucl Acids Res. 2004. 32: 537-41
24. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M. Global epidemiology of sickle cell hemoglobin in neonates: A contemporary geostatistical model-based map and population estimate. Lancet. 2013. 381: 142-51
25. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Eng J Med. 2017. 376: 1561-73
26. Rivierez M, Heyman D, Bazin M. Vertebral osteomyelitis with epidural abscess in a child with sickle cell disease. Neurochirurgie. 2000. 46: 47-9
27. Schnog JB, Duits AJ, Muskiet FA, ten Cate H, Rojer RA, Brandjes DP. Sickle cell disease: A general overview. Neth J Med. 2004. 62: 364-74
28. Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin variants: Biochemical properties and clinical correlates. Cold Spring Harb Perspect Med. 2013. 3: a011858-
29. Vichinsky EP, Neumayr LD, Haberkern C, Earles AN, Beckman J, Koshy M. The peri-operative complication rate of orthopaedic surgery in sickle cell disease: Report on the national sickle cell surgery study group. Am J Hematol. 1999. 62: 129-38
30. Weatherall D. The inherited disorders of hemoglobin: An increasingly neglected global health problem. Indian J Med Res. 2011. 134: 493-7
31. Wilkie DJ, Johnson B, Mack AK, Labotka R, Molokie RE. Sickle cell disease: An opportunity for palliative care across the life span. Nurs Clin North Am. 2010. 45: 375-97


